概述
骨髓增生异常综合征(myelodysplastic syndromes,MDS)是一组异质性后天性克隆性疾患,其基本病变是克隆性造血干、祖细胞发育异常(dysplasia),导致无效造血以及恶性转化危险性增高。表现为骨髓中各系造血细胞数量增多或正常,但看明显发育异常的形态改变;外周血中各系血细胞明显减少。而且演变为急性髓系白血病(acute myeloid leukemia,AML)的危险性很高。 对本综合征的认识经历了一个较长的过程,从20世纪40年代开始,文献中报道的白血病前期(preleukemia)、难治性贫血(refractory anemia)、难治性贫血伴有原始粒细胞过...[详细]
病因
MDS发病原因尚未明了,但从细胞培养、细胞遗传学、分子生物学及临床研究均证实,MDS是一种源于造血干/祖细胞水平的克隆性疾病。其发病原因与白血病类似。目前已经证明,至少2种淋巴细胞恶性增生性疾病——成人T细胞白血病及皮肤T细胞型淋巴瘤是由反转录病毒感染所致。亦有实验证明,MDS发病可能与反转录病毒作用或(和)细胞原癌基因突变、抑癌基因缺失或表达异常等因素有关。涉及MDS患者发病的常见原癌基因为N-ras基因。Ras基因家族分为H、N、K三种,MDS患者中最常见的为N-ras基因突变,发生在12、13、61外显子处,突变后N-ras基因编码蛋白表达异常,干扰了细胞正常增生和分化信号,导致细胞增生...[详细]
发病机制
MDS患者在致病因素作用下,引起患者造血干细胞损伤,用G6PD同工酶类型,X染色体伴限制性长度片段多态性甲基化,X染色体失活分析等方法已确定大部分MDS是病变发生在造血干细胞水平的克罗恩病,因而不但髓系、红系、巨核系细胞受累,淋巴细胞系亦受影响,导致T、B细胞数量和功能异常,临床表现为免疫缺陷或自身免疫性疾病。但在部分患者中其发病可仅局限在粒、红、巨核、巨噬祖细胞水平,仅有粒、红、巨核、巨噬细胞等受累而无淋巴细胞受累。 MDS发病具有阶段特性,可能与不同原癌基因和抑癌基因的变化有关。原癌基因活化包括基因过量表达、扩张、重排、易位、点突变等,抑癌基因变化包括等位基因丢失、缺失、重排、突...[详细]
临床表现
1.症状 MDS临床表现无特殊性,最常见的为缓慢进行性贫血症状。包括面色苍白、乏力、活动后心悸、气短,老年人贫血常使原有的慢性心、肺疾病加重。严重的粒细胞缺乏可降低患者的抵抗力,表现为反复发生的感染及发热。严重的血小板降低可致皮肤淤斑、鼻出血、牙龈出血及内脏出血。少数患者可有关节肿痛,发热、皮肤血管炎等症状,多伴有自身抗体,类似风湿病。 2.体征 MDS患者体征不典型。常为贫血所致面色苍白,血小板减少所致皮肤淤点、淤斑。肝脾肿大者约占10%左右。极少数患者可有淋巴结肿大和皮肤浸润,多为慢性粒单核细胞白血病(CMMoL)型患者。 3.特殊类型临床表现 (1)5q...[详细]
 (6)17p-综合征:17号染色体短臂缺失(17p-)可发生于5%左右的MDS患者。多数由于涉及17p的非平衡易位,亦可由于-17、iso(17q)或单纯17p-。17p-常合并其他染色体异常。抑癌基因p53定位于17p13。上述各种核型异常所造成的17p-,缺失区带可不完全相同,但都包括p53基因区带。而且70%左右的17p-综合征患者有p53基因失活,说明另一个等位p53基因也发生了突变。 17p-综合征的血液学突出表现为粒系细胞发育异常,外周血中性粒细胞有假性Pelger-Huet核异常和胞质中小空泡。这种改变也可见于骨髓中不成熟粒细胞。患者临床上对治疗反应差,预后不良。 (7)CMML:20世纪70年代初,Hurdle等和Meischer等首先报道CMML,认为它是一种慢性骨髓增殖性疾病(MPD),其特征为外周血白细胞数正常或增高,偶可有幼粒或幼红细胞,单核细胞>0.8×109/L。骨髓有核细胞增多,可有发育异常的形态表现,以粒系增殖为主,单核细胞亦增多。Ph染色体阴性,可有脾脏肿大。后来FAB协作组因其有血细胞发育异常的形态表现,将之纳入MDS作为一个亚型。但由于本病有明显的MPD特征,这种归类一直受到质疑。现在WHO分类方案中,将CMML改划人新增的MDS/MPD大类中,解决了这一长时间以来的争议。但确有一些MDS患者,外周血白细胞数无明显升高(<13×109/L),而单核细胞>1×109/L,临床上亦无肝脾肿大。骨髓中血细胞发育异常的形态表现十分明显。完全符合MDS特征。这类患者并不具备MPD的特征,显然不应作为CMML归入MDS/MPD中,而仍应诊断为MDS。至于是否需在MDS单列亚型,则有待商榷。 (8)aCML:本病表现类似Ph(+)CML,外周血白细胞数明显升高,有>10%的各阶段不成熟粒细胞。但与Ph(+)CML不同的是嗜碱粒细胞无明显增多,外周血和骨髓中血细胞发育异常的形态表现十分明显,而且常为三系发育异常。Ph染色体和bcr-abl融合基因均阴性。临床上对治疗CML的药物反应较差,病程进展较快,中位存活时间一般<2年。过去本病被诊断为Ph(+)CML,作为CML的一个变异型。WHO分类方案制订指导委员会和临床顾问委员会讨论后认为,本病临床过程并非慢性,使用aCML的病名容易引起误解,以为它是与Ph(+)CML有关系的慢性疾病,但又未能就改换一个新的病名达成一致。最后决定沿用aCML的病名,将之归入MDS/MPD大类之中。 关于Ph(+)CML、aCML、CMML的血液学表现鉴别要点如表2所示。
(6)17p-综合征:17号染色体短臂缺失(17p-)可发生于5%左右的MDS患者。多数由于涉及17p的非平衡易位,亦可由于-17、iso(17q)或单纯17p-。17p-常合并其他染色体异常。抑癌基因p53定位于17p13。上述各种核型异常所造成的17p-,缺失区带可不完全相同,但都包括p53基因区带。而且70%左右的17p-综合征患者有p53基因失活,说明另一个等位p53基因也发生了突变。 17p-综合征的血液学突出表现为粒系细胞发育异常,外周血中性粒细胞有假性Pelger-Huet核异常和胞质中小空泡。这种改变也可见于骨髓中不成熟粒细胞。患者临床上对治疗反应差,预后不良。 (7)CMML:20世纪70年代初,Hurdle等和Meischer等首先报道CMML,认为它是一种慢性骨髓增殖性疾病(MPD),其特征为外周血白细胞数正常或增高,偶可有幼粒或幼红细胞,单核细胞>0.8×109/L。骨髓有核细胞增多,可有发育异常的形态表现,以粒系增殖为主,单核细胞亦增多。Ph染色体阴性,可有脾脏肿大。后来FAB协作组因其有血细胞发育异常的形态表现,将之纳入MDS作为一个亚型。但由于本病有明显的MPD特征,这种归类一直受到质疑。现在WHO分类方案中,将CMML改划人新增的MDS/MPD大类中,解决了这一长时间以来的争议。但确有一些MDS患者,外周血白细胞数无明显升高(<13×109/L),而单核细胞>1×109/L,临床上亦无肝脾肿大。骨髓中血细胞发育异常的形态表现十分明显。完全符合MDS特征。这类患者并不具备MPD的特征,显然不应作为CMML归入MDS/MPD中,而仍应诊断为MDS。至于是否需在MDS单列亚型,则有待商榷。 (8)aCML:本病表现类似Ph(+)CML,外周血白细胞数明显升高,有>10%的各阶段不成熟粒细胞。但与Ph(+)CML不同的是嗜碱粒细胞无明显增多,外周血和骨髓中血细胞发育异常的形态表现十分明显,而且常为三系发育异常。Ph染色体和bcr-abl融合基因均阴性。临床上对治疗CML的药物反应较差,病程进展较快,中位存活时间一般<2年。过去本病被诊断为Ph(+)CML,作为CML的一个变异型。WHO分类方案制订指导委员会和临床顾问委员会讨论后认为,本病临床过程并非慢性,使用aCML的病名容易引起误解,以为它是与Ph(+)CML有关系的慢性疾病,但又未能就改换一个新的病名达成一致。最后决定沿用aCML的病名,将之归入MDS/MPD大类之中。 关于Ph(+)CML、aCML、CMML的血液学表现鉴别要点如表2所示。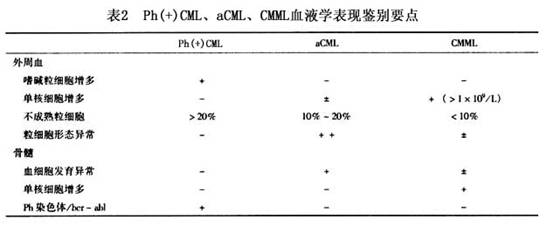
并发症
1.合并骨髓纤维化 近50%的MDS患者骨髓中有轻~中度网状纤维增多,其中:10%~15%的患者有明显纤维化。与原发性骨髓纤维化症不同的是,MDS合并骨髓纤维化者外周血常全血细胞减少,异形和破碎红细胞较少见;骨髓常示明显三系发育异常,胶原纤维形成十分少见。而且常无肝脾肿大。MDS合并骨髓纤维化可见于各个亚型,有作者认为是提示不良预后的因素之一。另有一种罕见的情况,称为急性骨髓增生异常伴有骨髓纤维化(acutemyelodysplasia with myelofibrosis,AMMF)。患者急性起病,有贫血、出血、感染等症状和体征,无肝脾肿大。外周血中全血细胞减少,成熟红细胞形态改变较轻,仅...[详细]
实验室检查
1.外周血 全血细胞减少是MDS患者最普遍也是最基本的表现。少数患者在病程早期可表现为贫血和白细胞或血小板减少。极少数患者可无贫血而只有白细胞和(或)血小板减少。但随着病程进展,绝大多数都发展为全血细胞减少。MDS患者各类细胞可有发育异常的形态改变。外周血可出现少数原始细胞、不成熟粒细胞或有核红细胞。 2.骨髓象 有核细胞增生程度增高或正常,原始细胞百分比正常或增高,红系细胞百分比明显增高,巨核细胞数目正常或增多,淋巴细胞百分比减低。红、粒、巨核系细胞至少一系有明确的上述发育异常的形态改变,常至少累及二系。 (1)红细胞生成异常(dyserythropoiesis):外...[详细]
其他辅助检查
1.病理检查 ①造血组织面积增大(>50%)或正常(30%~50%)。②造血细胞定位紊乱:红系细胞和巨核细胞不分布在中央窦周围,而分布在骨小梁旁区或小梁表面;粒系细胞不分布于骨小梁表面而分布在小梁间中心区,并有聚集成簇的现象。③(粒系)不成熟前体细胞异常定位(abnormal localization of immature precursors,ALIP)现象:原粒细胞和早幼粒细胞在小梁间中心区形成集丛(3~5个细胞)或集簇(>5个细胞)。每张骨髓切片上都能看到至少3个集丛和(或)集簇为ALIP( )。④基质改变:血窦壁变性、破裂,间质水肿,骨改建活动增强,网状纤维增多等。 2....[详细]
诊断
1.诊断标准 (1)发病原因分类:①原发性MDS,无明确发病原因;②继发性MDS,多继发于长期放、化疗后,亦可继发于自身免疫病、肿瘤等。 (2)形态学分类: ①法、美、英等国协作组分类(FAB分型)诊断标准: A.难治性贫血(RA):血象:贫血,偶有粒细胞减少、血小板减少而无贫血,网织红细胞减少。红细胞和粒细胞形态可有异常,原始细胞无或<1%;骨髓象:增生活跃或明显活跃。红系增生并有病态造血现象。很少见粒系及巨核系病态造血现象。原始细胞<5%。 B.环状铁粒幼细胞增多性难治性贫血(RAS):铁染色显示骨髓中环形铁粒幼细胞占所有有核细胞数的15%...[详细]
预后
MDS的病程大致有以下三种主要演变模式: 第一种模式,患者病情稳定,骨髓中原始细胞不增多或轻微增多,但不超过5%。随诊中从未发生白血病转变,仅靠一般支持治疗可存活数年甚至十多年。 第二种模式,患者初期病情稳定,与第一种相似,骨髓中原始细胞不增多或轻度增多,但一般<10%。经过一段时间以后,骨髓中原始细胞突然迅速增多,转变为AML。 第三种模式,患者骨髓中原始细胞缓渐地进行性增多,临床病情随之进展,直至转变为AML。 MDS患者骨髓细胞生物学特性的异常改变常提示发生白血病转变的可能性,如出现新的染色体异常或癌基因异常、细胞周期延长、体外培养呈现白血病样生长...[详细]
 对MDS诸多参数的预后意义进行分析的结果表明,最主要的预后因素是骨髓中原始细胞百分数,百分数愈高,预后愈差。染色体异常(尤其是-7/7q-、 8或复杂核型异常)也具有非常重要的意义。其他具有独立不良预后意义的因素尚有:外周血细胞显著减少,尤其是血小板减少和全血细胞减少,高龄(>60岁),ALIP( ),巨核细胞异常(特别是有淋巴细胞样小巨核细胞),伴有骨髓纤维化,SCD(-)等。 一些作者选取预后意义较强而又较易得到的几个参数,设计了MDS预后的积分系统。并通过较大病例系列的回顾性分析,证明积分高组的预后较积分低组为差。认为应用这些积分系统计算患者就诊时积分,对估计预后和决定治疗方针有一定帮助。表7为 MDS预后积分系统举例。
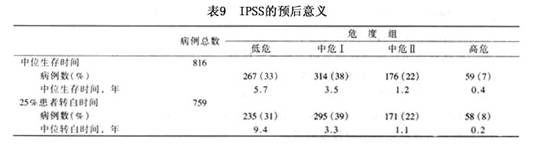
对MDS诸多参数的预后意义进行分析的结果表明,最主要的预后因素是骨髓中原始细胞百分数,百分数愈高,预后愈差。染色体异常(尤其是-7/7q-、 8或复杂核型异常)也具有非常重要的意义。其他具有独立不良预后意义的因素尚有:外周血细胞显著减少,尤其是血小板减少和全血细胞减少,高龄(>60岁),ALIP( ),巨核细胞异常(特别是有淋巴细胞样小巨核细胞),伴有骨髓纤维化,SCD(-)等。 一些作者选取预后意义较强而又较易得到的几个参数,设计了MDS预后的积分系统。并通过较大病例系列的回顾性分析,证明积分高组的预后较积分低组为差。认为应用这些积分系统计算患者就诊时积分,对估计预后和决定治疗方针有一定帮助。表7为 MDS预后积分系统举例。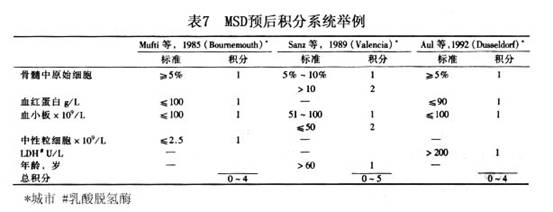 1997年国际MDS危险分析专题讨论会综合一些大系列的MDS预后资料,经过对各个重要预后因素的逐个分析,确定骨髓原始细胞%、骨髓造血细胞染色体核型和外周血细胞减少系列数最具有预后意义。据此提出一个MDS国际预后积分系统(International Prognostic Scoring System,IPSS)将MDS分为低危、中危Ⅰ、中危Ⅱ和高危四个危度组(表8),对提示患者的生存期及白血病转变具有肯定意义(表9)。IPSS提出后,很快得到一些作者的验证和认同,现已取代其他的预后积分系统,而被广泛接受。不少作者已将它视为一个提示预后和指导治疗的临床MDS分型方案。
1997年国际MDS危险分析专题讨论会综合一些大系列的MDS预后资料,经过对各个重要预后因素的逐个分析,确定骨髓原始细胞%、骨髓造血细胞染色体核型和外周血细胞减少系列数最具有预后意义。据此提出一个MDS国际预后积分系统(International Prognostic Scoring System,IPSS)将MDS分为低危、中危Ⅰ、中危Ⅱ和高危四个危度组(表8),对提示患者的生存期及白血病转变具有肯定意义(表9)。IPSS提出后,很快得到一些作者的验证和认同,现已取代其他的预后积分系统,而被广泛接受。不少作者已将它视为一个提示预后和指导治疗的临床MDS分型方案。
 MDS患者的死亡原因,约半数左右是由于骨髓无效造血加重,外周血中血细胞进行性减少而招致的出血和感染。30~40%是由于发生白血病转变。10%~20%是由于与MDS无直接关系的其他疾病。
MDS患者的死亡原因,约半数左右是由于骨髓无效造血加重,外周血中血细胞进行性减少而招致的出血和感染。30~40%是由于发生白血病转变。10%~20%是由于与MDS无直接关系的其他疾病。预防
继发性MDS常有明显的发病诱因,所以应避免或减少有害物质。如:放射线、化学物质、化学药物的接触。


