先天性巨结肠
该疾病知识由微医集团编辑审核

-
科室:
小儿普外科
-
别名:
HirschSprung disease
aganglionar Megacolon
赫什朋病
-
症状:
腹胀
腹痛
儿童偏食
-
发病部位:
暂无
-
多发人群:
婴幼儿人群
-
相关疾病:
先天性肠疾病
先天性巨结肠为一错误的命名,因为巨结肠改变不是先天性的。由于巨结肠的远端肠壁内没有神经节细胞,处于痉挛狭窄状态,丧失蠕动和排便功能,致使近端结肠蓄便、积气,而续发扩张、肥厚,逐渐形成了巨结肠改变。 1886年丹麦医生Harald Hirschsprung报道7个月和11个月2例病儿,详细描述了便秘症状和死后扩张结肠的肉眼所见,2年后该文章发表,所以也将该症称为赫什朋病(HirschSprung disease,HD)。由于他认为病变部位在巨结肠,故先天性巨结肠这一病名沿用至今。目前有些文献已将该病称为无神经节细胞症(aganglionosis),或无神经节细胞性巨结肠(aganglionar Megacolon,AM)。[收起]
先天性巨结肠为一错误的命名,因为巨结肠改变不是先天性的。由于巨结肠的远端肠壁内没有神经节细胞,处于痉挛狭窄状态,丧失蠕动和排便功能,致使近端结肠蓄便、积气,而续发扩张、肥厚,逐渐形成了巨结肠改变。 1886年丹麦医生Harald Hirschsprung报道7个月和11个月2例病儿,详细描述了便秘症状和死后扩张结肠的肉眼所见,2年后该文章发表,所以也将该症称为赫什朋病(HirschSprung disease,HD)。由于他认为病变部位在巨结肠,故先天性巨结肠这一病名沿用至今。目前有些文献已将该病称为无神经节细胞症(aganglionosis),或无神经节细胞性巨结肠(agangli...[详细]
1.胚胎学 Bodian认为,先天性巨结肠症的肠壁内神经节细胞缺如是一种壁内神经发育停顿,致使外胚层神经纤维无法参与正常的壁内神经丛发育。1954年Yntema和Hamman在胚胎研究发现,消化道的内在神经丛是由中枢神经嵴衍生而来。其神经母细胞沿已发育的迷走神经干迁移至整个消化道壁内,由头端之食管直至尾端之直肠,此即单相发育学说。而Tam等则提出神经节细胞系由口和肛门向中心发育,此即双相发育学说。 1967年Okamoto等对18例胚胎和胎儿进行了研究,发现肌间神经丛系由神经嵴的神经母细胞形成。这些神经母细胞于胚胎第5周开始沿迷走神经干由头侧向尾侧迁移,于第12周达到消化道远端。在胚胎第5周时已在食管壁发现神经母细胞,第6周至胃,第7周达中肠远端,第8周到横结肠中段,最后于12周布满全部消化道管壁至直肠。但是,直肠的末端即内括约肌神经母细胞尚未进入。在胚胎发育后期,肠壁内神经母细胞作为神经元,逐渐发育成为神经节细胞。不难设想,如果由于各类原因导致神经母细胞移行时中途停顿,即可造成肠壁无神经节细胞症。停顿的时间越早,则导致结肠远端无神经节细胞肠管越长。由于直肠、乙状结肠是在消化道的最远端,所以受累的机会最多(约85%)。神经母细胞由肌层向黏膜下发展,在纵肌与环肌形成肌间神经丛,即Auerbach神经丛。黏膜下的神经节细胞乃由肌间神经母细胞移行而来,穿过环行肌后,在黏膜下层形成黏膜深层神经丛,即Henley神经丛。神经母细胞再向内发展形成黏膜浅神经丛,即Meissner神经丛。临床上全层活检主要检查肌间神经丛,而吸引活检是主要检查黏膜下浅神经丛,即Meissner神经丛。国内王光大等对早产婴儿、新生儿、婴幼儿的结肠、直肠肌间神经丛和黏膜下神经丛(包括深层、浅层)神经节细胞进行了研究,其结果也支持上述学说。 近来Okamoto用嗜银染色法检查,发现先天性巨结肠病儿神经节细胞缺如仅限于肠壁,而同属盆丛神经支配的膀胱、前列腺等神经节细胞均为正常。这不仅表现在外来自主神经纤维和自主神经感觉健在,而且其排列结构均无异常。上述结果说明,先天性巨结肠症的病理改变源于肠壁本身,并非因盆丛的原发病变所引起。研究资料还证实盆丛神经(副交感神经)原基在胚胎6周时已经形成,其神经母细胞迂回于直肠周围到膀胱左右基底部,约于第8周时形成膀胱、前列腺(子宫)神经丛,这时尚未见到有明显的分支及神经母细胞进入直肠。直肠壁内神经丛形成比盆丛稍晚,约在第10周以后,然后由盆丛的副交感神经纤维移入结肠直肠壁内与沿消化道迁移来的神经节会合形成肠壁肌间神经丛。如果无肠壁内神经节细胞,则盆丛的副交感神经纤维必定在肌间大量增生,此即病变肠段重要的病理改变之一。如果盆丛发生病变,则肌间神经丛也不可能正常发育,两者相辅相成。近端结肠的副交感神经系来自迷走神经。在全结肠型病例中副交感神经纤维有时减少或缺如 2.遗传学因素 Valle(1924)首先发现先天性巨结肠有家族遗传性,此后关于先天性巨结肠的家族性发病报道逐渐增多。随着遗传学的深入研究,认识到先天性巨结肠是遗传与环境因素的联合致病作用,为多基因或多因素遗传病,也有人称之为性修饰多因素遗传病(sex-modified multifactorial inhertitance),遗传度为80%。分子遗传学用于先天性巨结肠的病因学研究后,目前已发现5个突变基因:RET基因、GDNF基因、EDN3基因、EDNRB基因和SOX10基因。 (1)RET原癌基因(proto-oncogene RET):Takahashi与Cooper(1987)年在重组DNA的实验中,首次发现RET原癌基因。RET基因定位于10q11.2区。Martucciello(1992)报道1例10号染色体长臂(10q)缺失的全结肠型先天性巨结肠女性病儿。现已确定,DNA全长约8万个核苷酸(80Kb),有21个外显子,至少有4个转录产物,且在不同的组织中含量不同。RET蛋白为1114个残基跨膜蛋白,有一个富含半胱氨酸的钙粘连素样细胞外区,一个跨膜区和一个催化酪氨酸激酶(TK)的细胞内区。TK受体(TKR)的基本功能是,将细胞外信息转变为可传入细胞内的化学信号。Tahira等(1988)研究小鼠组织中RET的mRNA表达情况:成鼠组织中未能查出RET的mRNA表达,而胚胎鼠的中枢及外周神经系统(包括肠内神经系统)却能查到。Pachnis等(1993)发现,当RET表达量减少一半时,神经节细胞就不能移行到肠壁内。说明RET对肠内神经系统的发育起重要作用。现已证实RET基因突变是引起先天性巨结肠的主要基因,50%家族性先天性巨结肠、7.3%~20%散发性先天性巨结肠,与RET、基因突变有关。 (2)胶质细胞源性神经营养因子(glia[收起]
1.胚胎学 Bodian认为,先天性巨结肠症的肠壁内神经节细胞缺如是一种壁内神经发育停顿,致使外胚层神经纤维无法参与正常的壁内神经丛发育。1954年Yntema和Hamman在胚胎研究发现,消化道的内在神经丛是由中枢神经嵴衍生而来。其神经母细胞沿已发育的迷走神经干迁移至整个消化道壁内,由头端之食管直至尾端之直肠,此即单相发育学说。而Tam等则提出神经节细胞系由口和肛门向中心发育,此即双相发育学说。 1967年Okamoto等对18例胚胎和胎儿进行了研究,发现肌间神经丛系由神经嵴的神经母细胞形成。这些神经母细胞于胚胎第5周开始沿迷走神经干由头侧向尾侧迁移,于第12周达到消化道远端。在...[详细]
1.病理及神经免疫组化改变 先天性巨结肠症的受累肠段可以见到典型的改变,即明显的狭窄段和扩张段。狭窄段位于扩张段远端,一般位于直肠乙状结肠交界处以远,距肛门7~10cm以内。狭窄肠管细小,与扩大肠管直径相差悬殊,其表面结构无甚差异。在与扩大结肠连接部形成漏斗状的移行区(即扩张段远端移行区),此区原属狭窄段,由于近端肠管的蠕动,推挤肠内容物向前移动,长期的挤压促使狭窄段近端肠管扩大成漏斗形。扩张段多位于乙状结肠,严重者可波及横结肠。该肠管异常扩大,其直径较正常增大2~3倍,最大者可达10cm以上。肠壁肥厚、质地坚韧如皮革状。肠管表面失去红润光泽,略呈苍白。结肠带变宽而肌纹呈纵行条状被分裂。结肠袋消失,肠蠕动极少。肠腔内含有大量积粪,偶能触及粪石。切开肠壁见原有的环形肌、纵形肌失去正常比例(2.2∶1),甚至出现比例倒置。肠壁厚度为狭窄段2倍,肠黏膜水肿、光亮、充血而粗糙,触之易出血,有时可见有浅表性溃疡。先天性巨结肠症的主要病理改变位于扩张段远端的狭窄肠管。狭窄段肌间神经丛(Auerbach丛)和黏膜下神经丛(Meissner丛)内神经节细胞缺如,其远段很难找到神经丛。神经纤维增粗,数目增多,排列呈波浪状。有时虽然找到个别的神经节细胞,形态亦不正常。狭窄段近端结肠壁内逐渐发现正常神经丛,神经节细胞也渐渐增多。黏膜腺体呈不同程度的病损,结肠固有膜增宽,并伴有淋巴细胞、嗜伊红细胞、浆细胞和巨噬细胞浸润,有时可见浅表性溃疡。 HD是直肠或结肠某段肠神经系统(enteric nervous system,ENS)神经元的发育异常,根据我们的实验研究其突出的神经病理改变是肠狭窄段壁内神经丛,各种ENS神经元均缺失(无神经节细胞肠段)或显著缺少或发育异常(少神经节细胞肠段)。肠肌间神经丛(Auerbach丛)和黏膜下神经丛(Meissener丛)的神经突触网络联系也发生相应的改变。同时,各类外源性神经的支配发生广泛的紊乱。①肠壁各层副交感胆碱能神经节前纤维异常增生、增粗、酶活性增强,固有膜内出现乙酰胆碱酶(acetylcholinesterase,AChE)阳性神经(代表胆碱能神经),具有特征性改变,且与临床症状(痉挛梗阻程度)相关,可作为诊断本症的重要依据。②壁内含去甲肾上腺素(noradrenaline,NA)荧光的交感节后纤维也同样增多、增粗,其特征是缺失正常的肠壁神经丛周围的篮样丛神经突触网络结构。③肽能神经支配广泛的紊乱;壁内丛缺失任何肽能神经元;肠壁各层P物质(substance P,SP)、脑啡肽(enkephalin,ENK)诸纤维减少,肠肌丛部位(肌间隙或肌束间)和黏膜层则出现大量增粗的含血管活性肠肽(vasoactive intestinal polypeptide,VIP)、降钙素基因相关肽(calcitonin gene releted peptide,CGRP)、生长抑素(somatostatin,SDM)和神经肽Y(neuropeptide Y,NPY)诸纤维束或小神经干或纤维网,NPY支配过盛更明显。但黏膜下血管中后4种肽能神经明显减少或缺失。④含一氧化氮(nitric oxide,NO)神经成分在无神经节细胞结肠段的改变,与VIP相似。⑤含5-羟色胺(serotonin或5-hydroxytryptamine,5-TH)能神经元成分,在无神经节细胞的肠肌丛周的终末网及肌层内的神经纤维束网明显减少。本病狭窄段结肠内上述多种神经成分的改变,主要累及运动神经元,对黏膜的分泌和感觉的神经支配也有影响,可能是导致乙酰胆碱(acetyl-choline,ACh)、SP、ENK、CGRP和NPY(兴奋性神经递质,刺激肠肌收缩)与NA、VIP、SOM和NO(抑制性神经递质,抑制肠肌运动)参与调节结肠运动动力功能失控,成为本病无神经节细胞狭窄肠段痉挛收缩的神经化学病理生理诸多因素。 2.病理生理 结肠和内括约肌的运动机制非常复杂,传统的概念认为其神经支配为交感神经和副交感神经。前者使平滑肌抑制,即松弛作用;后者使平滑肌兴奋,即收缩作用。而在内括约肌两者作用相反。结肠壁内神经节被认为是副交感神经系统。近年来,通过临床病理、组织化学、电子显微镜检查、药物反应试验以及动物实验等手段,了解到结肠及内括约肌的神经支配共分3部分,已如前述。 先天性巨结肠症的病理改变是由于狭窄肠段无神经节细胞,冈本英三(1988)研究证实在病变肠段未找到神经与肌肉的连接点(缺如),并在神经递质受体定量测定时,发现无论是胆碱能受体或肾上腺能β受体的含量均较正常肠段明显减少,从而造成病变肠管及内括约肌痉挛狭窄和缺乏正常的蠕动功能,形成功能性肠梗阻。本应与神经节细胞建立突触联系的副交感神经节前纤维在无神经节细胞肠段大量增生变粗,交感神经节后纤维亦明显增多。大量释放乙酰胆碱被认为是引起肠段痉挛的主要原因之一,胆碱能神经节细胞缺乏后,阻断了正常的节段性运动和节律性推进蠕动。而来自骶部副交感神经又直接作用于肠壁肌细胞,因而使病变肠管产生持续性强直收缩。此外,也由于神经节细胞缺如,增生的交感神经中断原有的抑制通路,不能由β抑制受体去影响胆碱能神经,从而产生肠壁松弛,而是直接到达平滑肌的α兴奋受体产生痉挛。壁内的非胆碱能非肾上腺素能系统抑制神经元也缺乏,因而失去有效的松弛功能。由于直肠、内括约肌保持在持续性收缩状态,导致肠道的正常推进波受阻。最后形成粪便潴留、腹胀、大便不能排出。检查时可见结肠正常蠕动波不能下传。无神经节细胞肠管不但缺乏神经节细胞,特别是交感能神经的数目也为之减少,这种几乎完全处于无神经支配的状态(Cannon定律),导致肠管强直性挛缩。久之,近端正常肠段疲惫不堪,发生代偿性、继发性扩大肥厚,神经节细胞亦产生退化变性直至萎缩,以致减少或消失。 Swenson将气囊放入结肠,记录各段肠管的蠕动,发现正常儿肠管与病变肠段完全不同,前者当肠蠕动进入乙状结肠时收缩明显增加;而在无神经节细胞肠管,当肠蠕动传至乙状结肠时并无收缩波出现,这一现象当可解释患儿的便秘与梗阻症状。随着病儿年龄的增大,肠管愈加扩大,便秘进行性加重,而继发性病变肠段更趋延长,以致波及近端结肠或小肠。 这种长期慢性梗阻的结果必然导致患儿食欲不佳,营养吸收障碍,生长发育差,贫血、低蛋白血症等。肠内大量细菌繁殖造成菌群失调后,毒素吸收又将引起心、肝、肾功能受损,最后因抵抗力低下感染衰竭或肠炎穿孔而死亡。[收起]
1.病理及神经免疫组化改变 先天性巨结肠症的受累肠段可以见到典型的改变,即明显的狭窄段和扩张段。狭窄段位于扩张段远端,一般位于直肠乙状结肠交界处以远,距肛门7~10cm以内。狭窄肠管细小,与扩大肠管直径相差悬殊,其表面结构无甚差异。在与扩大结肠连接部形成漏斗状的移行区(即扩张段远端移行区),此区原属狭窄段,由于近端肠管的蠕动,推挤肠内容物向前移动,长期的挤压促使狭窄段近端肠管扩大成漏斗形。扩张段多位于乙状结肠,严重者可波及横结肠。该肠管异常扩大,其直径较正常增大2~3倍,最大者可达10cm以上。肠壁肥厚、质地坚韧如皮革状。肠管表面失去红润光泽,略呈苍白。结肠带变宽而肌纹呈纵行条状被分裂。结肠...[详细]
1.新生儿和婴幼儿 正常新生儿几乎全部在生后24h内排出第一次胎便。患儿则表现为生后不排胎便,开始排胎便及排空时间均推迟。生后2~3天内出现部分甚至完全性肠梗阻症状。患儿呕吐,吐物含胆汁或粪便样液体,并有腹胀及便秘。肛门指诊或用温盐水洗肠,可排出大量胎便及气体,症状缓解。缓解数天后,腹胀、便秘又复出现,又需洗肠才能排便。体检可完全正常,有时可扪到扩张的肠管横贯于上腹部。如果看到有从右向左的蠕动波,可以推测是在无神经节细胞肠段近端扩张的横结肠。腹胀缓解后可以进食,惟多数患儿由于呕吐、拒食而体重不增,发育较差。 在新生儿,也可能以腹泻为突出症状而伴有肠梗阻,经常便秘与腹泻交替出现。如反复迁延,患儿日趋消瘦,并引起营养不良性水肿。新生儿期,并发肠炎者甚为多见,炎症往往顽固难治,可发展为凶猛的小肠结肠炎。发病较急,有高热,吐、泻,梗阻肠腔内积存的大量肠液可导致重度脱水、酸中毒和休克,如不积极抢救,多于24h内死亡。肠炎病变可产生腹腔渗液,出现腹膜刺激征,临床上很像化脓性腹膜炎。 如症状较轻,于新生儿期往往未能作出诊断。到婴幼儿期表现为进行性便秘及腹胀。轻症者常需泻药通便,重症者每需洗肠协助排便。患儿发育营养均低于正常标准,腹胀明显,腹壁静脉曲张,常出现肠型,左下腹部可触及充满粪便的肠袢。 2.年长儿和成人 有些年长儿和成人对此病耐受性好,可以活到较大年龄。顽固性便秘是主要的病史,有的2周才解一次大便,经常依靠泻药排便,而且泻药越来越不起作用。体检可见面色苍白,营养不良,腹部突出,两腿细长。偶见急性肠功能不全。病人发展到完全性肠梗阻而就医。Meyer报道一例37岁男性病人,每天必须服3两矿物油,最后发生盲肠憩室穿孔,其神经节细胞缺如一直达到横结肠的水平。偶见无神经节细胞肠段只限于肛门括约肌的内侧(超短型巨结肠病),患者可有大便失禁。这种情况除非做特别低位的肠活检来证实,否则因找见神经节细胞而误诊为正常。[收起]
1.新生儿和婴幼儿 正常新生儿几乎全部在生后24h内排出第一次胎便。患儿则表现为生后不排胎便,开始排胎便及排空时间均推迟。生后2~3天内出现部分甚至完全性肠梗阻症状。患儿呕吐,吐物含胆汁或粪便样液体,并有腹胀及便秘。肛门指诊或用温盐水洗肠,可排出大量胎便及气体,症状缓解。缓解数天后,腹胀、便秘又复出现,又需洗肠才能排便。体检可完全正常,有时可扪到扩张的肠管横贯于上腹部。如果看到有从右向左的蠕动波,可以推测是在无神经节细胞肠段近端扩张的横结肠。腹胀缓解后可以进食,惟多数患儿由于呕吐、拒食而体重不增,发育较差。 在新生儿,也可能以腹泻为突出症状而伴有肠梗阻,经常便秘与腹泻交替出现。如反复...[详细]
小肠结肠炎及肠穿孔是先天性巨结肠的常见并发症,亦是引起死亡最多见的原因。有文献统计有20%~50%的病儿并发小肠结肠炎,其病死率约30%。肠炎可以发生在各种年龄,但以3个月以内婴儿发病率最高。90%的肠炎病例发生于2岁以内,以后逐渐减少。即使在根治术后或结肠造瘘术后亦偶有出现结肠炎者。Shono报道术后发生肠炎者占61%而多见于Boley手术后。Soaue报道Sweorson术后肠炎占11.6%。Ikeda报道术后肠炎占33.7%,Soalce术后为19.5%,Boley术后12.1%。因此术后预防治疗肠炎成为重要课题。有作者统计先行造瘘术而后发生肠炎者,病死率可以降低。引起肠炎的原因和机制至今尚不十分明了,近10年来对其疗效也无显著改进。许多学者提出小肠结肠炎可能有以下几个原因: 1.肠梗阻 Swenson最早提出肠炎是由于梗阻所致。无神经节细胞肠管痉挛狭窄,缺乏蠕动功能,因而促使肠炎发生。所以国外均主张HD一经诊断立即造瘘,但这一理论不能解释造瘘术后梗阻已经解除仍有肠炎发生。 2.细菌毒素 巨结肠病人大便潴留,细菌大量繁殖,菌群失调。1986年Thorns等用梭状芽孢杆菌抗血清法,检查13例合并肠炎患儿,其中54%有细菌毒素存在。而非巨结肠排稀便者12例,仅1例毒素阳性。并且在此13例患儿粪便中分离出梭状芽孢杆菌10例占77%,这些结果均说明梭状芽孢杆菌与肠炎产生有密切关系。由于细菌毒素的侵袭肠壁血管,使血管通透性增加,大量液体渗出流入肠腔,造成水泻、腹胀。毒素吸收后出现高热(39~40℃),病儿进而产生败血症、休克衰竭、DIC、肠穿孔等而死亡。 3.过敏反应 HD小肠结肠炎,无论手术与否均可发生。常常病情凶猛、发展迅速。有的患儿即使一直住在医院进行细心的洗肠补液,甚至术后亦可突然发病而死亡。所以有学者指出,这些患儿是由于肠黏膜对某些细菌抗原有超敏反应,加之细菌侵入而发生败血症死亡。 4.局部免疫功能低下 肠黏膜屏障由3层保护层组成:①细胞前保护层:主要由杯状细胞分泌黏液所形成的一道物理屏障及正常菌丛形成的微生物屏障和分泌型lgA形成的保护膜;②肠细胞保护层:由肠细胞及多糖蛋白复合物构成;③细胞后保护层:由细胞下结缔组织、毛细血管和淋巴管共同构成。近年来有人提出小肠结肠炎系局部免疫损害所致。金子十郎研究证实巨结肠严重肠炎时,结肠局部产生IgA细胞数目和分泌量均明显减少和降低。肠壁的IgA系统也有下降趋势。免疫球蛋白IgA在肠道中起着一种天然的保护膜作用,双体IgA才能结合补体,固着于革兰阴性杆菌后,被IgA所活化的补体系统使溶菌酶能消化细菌包壁上的黏多糖。单体IgA亦能通过淋巴管从固有层进入血流,在肠道感染时可以使血清中的IgA增高。巨结肠发生肠炎时破坏了正常的免疫反应,因而导致肠炎反复发作。这些患儿抵抗力低下也容易发生上呼吸道感染。有人在研究PL鼠肠炎时亦发现患鼠局部免疫球蛋白产生细胞明显低于对照组,同时发现中性黏蛋白及磷黏蛋白耗尽而且杯状细胞有丝分裂活动很低。病鼠缺乏磷酸盐可能导致对细菌的敏感性。Teitelbaum亦报道患鼠在发病时局部免疫球蛋白Ig和白蛋白均明显下降,上述研究结果均可提示肠炎的发生与局部免疫有关,然而这些局部免疫的缺陷是原发而导致肠炎的发生,抑或继发于肠炎尚有待进一步证明。 肠炎发生时进行结肠镜检查,可以见到黏膜水肿、充血以及局限性黏膜破坏和小型溃疡。轻擦也容易出血。病变加重时向肌层发展,出现肠壁全层水肿、充血、增厚,在巨大病灶的浆膜层可见有黄色纤维膜覆盖。如病变进一步发展即可发生肠穿孔,并导致弥漫性腹膜炎。其病理检查可见隐窝脓肿、变性、绒毛炎性细胞浸润以及淋巴滤泡增生。1994年Kobayashi用单抗检测细胞内黏分子(ICAM-1),以了解其在HD合并肠炎中的作用,结果发现肠炎时黏膜下血管上皮均可见到明显着色,而对照组则很少见到。ICAM-1能诱导炎症时许多组织的白细胞浸润,且诱导各种细胞出现炎性激素,如干扰素、白细胞介素-1及肿瘤坏死因子,它在白细胞的黏着及调节血管外白细胞起着重要作用,因此即使在肠炎发作间隙或未出现前,如果ICAM-1显色表明有肠炎发生的危险。 有严重肠炎时,患儿有频繁呕吐、水样腹泻、高热和病情突然恶化。腹部异常膨胀并呈现脱水症状。进而发生呼吸困难、衰竭、全身反应极差。少数病儿虽未出现腹泻,当进行肛门指检或插入肛管时迅即见有大量奇臭粪水及气体溢出。腹胀可随之消减,但不久又行加重。小肠结肠炎往往病情凶险,治疗若不及时或不适当可导致死亡。 由于肠炎时肠腔扩张,肠壁变薄缺血,肠黏膜在细菌和毒素的作用下产生溃疡、出血甚至穿孔形成腹膜炎,肠炎并发肠穿孔病死率更高,尤其是新生儿,可高达70%~80%。 5.水中毒 水中毒多见于乳幼儿。此外,新生儿期用大量低渗盐水洗肠,输液过量或过快;年长儿先天性巨结肠,伴有慢性营养不良、低蛋白血症,常有细胞性或间质性水肿,在洗肠或输液不当时,也容易发生水中毒。急性水中毒主要累及脑、心、肺。脑水肿症状为恶心、呕吐、昏迷或抽搐;也可出现心力衰竭或肺水肿。为预防水中毒,要严格控制输液量;洗肠时用等渗盐水,不能用肥皂水等低渗液体;要用虹吸法,不得用灌肠法洗肠。[收起]
小肠结肠炎及肠穿孔是先天性巨结肠的常见并发症,亦是引起死亡最多见的原因。有文献统计有20%~50%的病儿并发小肠结肠炎,其病死率约30%。肠炎可以发生在各种年龄,但以3个月以内婴儿发病率最高。90%的肠炎病例发生于2岁以内,以后逐渐减少。即使在根治术后或结肠造瘘术后亦偶有出现结肠炎者。Shono报道术后发生肠炎者占61%而多见于Boley手术后。Soaue报道Sweorson术后肠炎占11.6%。Ikeda报道术后肠炎占33.7%,Soalce术后为19.5%,Boley术后12.1%。因此术后预防治疗肠炎成为重要课题。有作者统计先行造瘘术而后发生肠炎者,病死率可以降低。引起肠炎的原因和机制至...[详细]
1.直肠肌层活检 Swenson(1955)最先采用该法,准确率为98%。从直肠壁取肌层活检,证实肌间神经节细胞缺如诊断先天性巨结肠,理论上是最可靠的方法,但因存在一些缺点,故目前并非必要:①Smith(1968)经组织学检查发现,生后神经节细胞有一发育和成熟过程,直肠肌间丛尤其明显,黏膜下丛又落后几周,如不注意,可把正常儿诊断为先天性巨结肠或同源病。②正常直肠在齿状线上方有一低神经节细胞区,在该区内取材可把正常儿诊断为先天性巨结肠或同源病。故强调取材高度在齿状线上方至少新生儿2cm,1岁以内2.5cm,1~3岁3cm,4岁以上3.5cm,如此,短段型巨结肠病儿又易漏诊。③小儿肌层活检有肠穿孔、出血、感染等并发症,术后瘢痕可影响根治性手术。新生儿因肛管狭小、直肠壁薄、操作不便等,更易发生并发症。 另外,肌层活检是诊断先天性巨结肠同源病的主要根据。 2.直肠黏膜活检 直肠黏膜活检仅吸取一小块黏膜。经不断改进,该法简单、安全可靠,可不需麻醉,不必住院。检查方法有组织学、组织化学及免疫组织化学。组织学检查主要用HE染色判断神经丛中神经节细胞的有或无。该法简单,但不准确;乙酰胆碱酯酶等组织化学检查方法,需要新鲜组织标本和冷冻切片机等技术条件,新生儿期因乙酰胆碱酯酶活性较低,易出现假阴性结果;免疫组织化学方法准确性高,但因试剂昂贵,目前尚不适于做常规诊断方法。王怡平等(1991)采用神经元特异性稀醇化酶(NSE)免疫组化法,检查32例怀疑先天性巨结肠病儿,无一例误诊;徐本源等(1995)用NSE和S-100蛋白,诊断先天性巨结肠,准确率达100%。NSE为神经节细胞内酶之一,是神经节细胞的标志,NSE抗体与神经节细胞及神经纤维内NSE结合,使神经组织清晰可见。发育未成熟的神经节细胞也容易辨认,并容易与炎性细胞、淋巴细胞、施万细胞、巨噬细胞和血管内皮细胞等区别;S-100蛋白是一种神经系统周围成分的标记物。应用NSE和S-100蛋白两种组化方法,异途同归、互为佐证,尤其是对新生儿期先天性巨结肠,有重要诊断价值。我们(1998)应用免疫组化技术,检测30例先天性巨结肠的无神经节细胞段和正常段肠管中P75NGFR的分布情况,认为该法操作简单、结果准确,可用于诊断先天性巨结肠。目前,同济医科大学、上海第二医科大学及中国医科大学都开展了组织化学及免疫组化检查。[收起]
1.直肠肌层活检 Swenson(1955)最先采用该法,准确率为98%。从直肠壁取肌层活检,证实肌间神经节细胞缺如诊断先天性巨结肠,理论上是最可靠的方法,但因存在一些缺点,故目前并非必要:①Smith(1968)经组织学检查发现,生后神经节细胞有一发育和成熟过程,直肠肌间丛尤其明显,黏膜下丛又落后几周,如不注意,可把正常儿诊断为先天性巨结肠或同源病。②正常直肠在齿状线上方有一低神经节细胞区,在该区内取材可把正常儿诊断为先天性巨结肠或同源病。故强调取材高度在齿状线上方至少新生儿2cm,1岁以内2.5cm,1~3岁3cm,4岁以上3.5cm,如此,短段型巨结肠病儿又易漏诊。③小儿肌层活检有肠穿...[详细]
1. X线钡剂灌肠 X线下钡剂灌肠是判定病变范围和选择术式的重要依据。钡剂灌肠目的是显示痉挛段及其上方的扩张段,因此确认扩张段即可,不要过多灌入钡剂继续向上检查,以免加重病儿腹胀及其危险。 痉挛段范围在降结肠以下者,侧位显示最清,故一般仅摄带肛门标记的侧位X线片,但痉挛段达乙状结肠时从正位观察才能全面(图1)。X线钡剂灌肠的诊断率,目前仍徘徊在90%左右,其原因主要有3个:①新生儿巨结肠确诊困难,一般认为新生儿巨结肠的形态学改变,生后2周才形成,有的需要3~4周甚至几个月。尽管开展了灌肠后24~48h,动态观察钡剂贮留或排泄的功能性改变,但梗阻症状很重时,钡剂灌肠后必须洗肠或手术,不允许延迟观察;并发肠炎时,难于保留钡剂达24h以上。②对短段型先天性巨结肠,尤其是超短段型先天性巨结肠,难与特发性巨结肠鉴别。③对特殊型先天性巨结肠易于漏诊或误诊。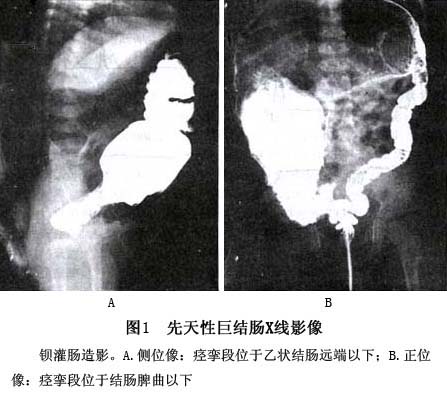 2.直肠肛管测压 是诊断先天性巨结肠的有效方法,具有经济、简便、快速而安全,以及无损伤性,可反复检测等优点。关于测压诊断先天性巨结肠的准确率,文献报道不一(76%~100%)。研究证明,正常儿直肠内气囊注入2~3ml气体后,1~3s内肛管压力迅速下降(称正常反射),而先天性巨结肠病儿,向直肠内气囊注入很多气体,肛管压力都不变(称阴性反射),即无直肠肛管反射或无正常反射,有的先天性巨结肠病儿,肛管压力不但不下降,反而上升(称异常反 射)(图2)。阴性反射和异常反射统称为病理反射,经检测156例慢性便秘病儿,直肠肛管测压的准确率为93.33%,其中误诊率为2.88%(104例病理反射者,3例除外先天性巨结肠:2例为新生儿,6个月后复查正常;另1例为胎粪性腹膜炎),漏诊率为7.69%(52例有正常反射者,4例为先天性巨结肠病儿)。为提高测压诊断的准确性,必须注意检测方法和判断标准。
2.直肠肛管测压 是诊断先天性巨结肠的有效方法,具有经济、简便、快速而安全,以及无损伤性,可反复检测等优点。关于测压诊断先天性巨结肠的准确率,文献报道不一(76%~100%)。研究证明,正常儿直肠内气囊注入2~3ml气体后,1~3s内肛管压力迅速下降(称正常反射),而先天性巨结肠病儿,向直肠内气囊注入很多气体,肛管压力都不变(称阴性反射),即无直肠肛管反射或无正常反射,有的先天性巨结肠病儿,肛管压力不但不下降,反而上升(称异常反 射)(图2)。阴性反射和异常反射统称为病理反射,经检测156例慢性便秘病儿,直肠肛管测压的准确率为93.33%,其中误诊率为2.88%(104例病理反射者,3例除外先天性巨结肠:2例为新生儿,6个月后复查正常;另1例为胎粪性腹膜炎),漏诊率为7.69%(52例有正常反射者,4例为先天性巨结肠病儿)。为提高测压诊断的准确性,必须注意检测方法和判断标准。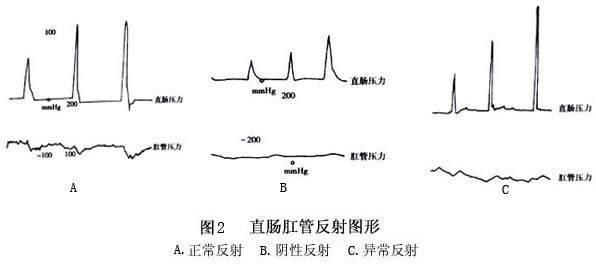 直肠肛管测压诊断新生儿巨结肠应当慎重。有作者动态检测50例正常新生儿,仅13例于生后第1天出现正常反射,48例(96%)在生后1周内出现正常反射,另2例因出院未能连续检测,分别在生后100天和8个月测检时出现正常反射。理论上应该说,新生儿生后自动排便,标志有直肠肛管反射,但经测压观察,我们认为这种刚刚形成或建立的反射,并未成熟也不稳定,故测检中不易显示或捕捉。 目前一致认为,诊断和鉴别超短段先天性巨结肠与特发性巨结肠的最可靠方法,是直肠肛管测压检查。[收起]
1. X线钡剂灌肠 X线下钡剂灌肠是判定病变范围和选择术式的重要依据。钡剂灌肠目的是显示痉挛段及其上方的扩张段,因此确认扩张段即可,不要过多灌入钡剂继续向上检查,以免加重病儿腹胀及其危险。 痉挛段范围在降结肠以下者,侧位显示最清,故一般仅摄带肛门标记的侧位X线片,但痉挛段达乙状结肠时从正位观察才能全面(图1)。X线钡剂灌肠的诊断率,目前仍徘徊在90%左右,其原因主要有3个:①新生儿巨结肠确诊困难,一般认为新生儿巨结肠的形态学改变,生后2周才形成,有的需要3~4周甚至几个月。尽管开展了灌肠后24~48h,动态观察钡剂贮留或排泄的功能性改变,但梗阻症状很重时,钡剂灌肠后必须洗肠或手术,...[详细]
直肠肛管测压诊断新生儿巨结肠应当慎重。有作者动态检测50例正常新生儿,仅13例于生后第1天出现正常反射,48例(96%)在生后1周内出现正常反射,另2例因出院未能连续检测,分别在生后100天和8个月测检时出现正常反射。理论上应该说,新生儿生后自动排便,标志有直肠肛管反射,但经测压观察,我们认为这种刚刚形成或建立的反射,并未成熟也不稳定,故测检中不易显示或捕捉。 目前一致认为,诊断和鉴别超短段先天性巨结肠与特发性巨结肠的最可靠方法,是直肠肛管测压检查。[收起]
1. X线钡剂灌肠 X线下钡剂灌肠是判定病变范围和选择术式的重要依据。钡剂灌肠目的是显示痉挛段及其上方的扩张段,因此确认扩张段即可,不要过多灌入钡剂继续向上检查,以免加重病儿腹胀及其危险。 痉挛段范围在降结肠以下者,侧位显示最清,故一般仅摄带肛门标记的侧位X线片,但痉挛段达乙状结肠时从正位观察才能全面(图1)。X线钡剂灌肠的诊断率,目前仍徘徊在90%左右,其原因主要有3个:①新生儿巨结肠确诊困难,一般认为新生儿巨结肠的形态学改变,生后2周才形成,有的需要3~4周甚至几个月。尽管开展了灌肠后24~48h,动态观察钡剂贮留或排泄的功能性改变,但梗阻症状很重时,钡剂灌肠后必须洗肠或手术,...[详细]
诊断先天性巨结肠,主要根据临床表现,确诊则需要X线钡剂灌肠、直肠肛管测压、直肠活检、组织化学等客观检查方法。 新生儿巨结肠的症状需要与胎粪性便秘、肠闭锁或狭窄、肛门直肠畸形等病鉴别。用小指肛诊后,巨结肠病儿能大量排出胎便及气体,随之症状缓解。因此,对新生儿肠梗阻,常规做肛诊检查,不仅有助于诊断和治疗新生儿巨结肠,而且可避免或减少错误诊治。 6个月以上病儿,有慢性便秘等症状、又有新生儿巨结肠史,诊断先天性巨结肠较容易,否则须除外肛门直肠畸形等病所致续发性巨结肠,以及克汀病、饮食性便秘、特发性巨结肠等症。肛诊时,短段型先天性巨结肠的肛门紧缩,直肠明显扩张积便或积气;普通型先天性巨结肠的直肠壶腹消失,处于紧缩(不是狭窄)状态,但这种状态并不恒定,直肠可因积存粪便而被动扩张。故不能根据肛诊检查结果确诊先天性巨结肠及其分型。另外,肛诊时还要注意先天性巨结肠病儿粪便的气味、性状等特点。 再次强调,对先天性巨结肠病儿必须做肛诊检查,一是有助于诊断,二是有利于鉴别。[收起]
诊断先天性巨结肠,主要根据临床表现,确诊则需要X线钡剂灌肠、直肠肛管测压、直肠活检、组织化学等客观检查方法。 新生儿巨结肠的症状需要与胎粪性便秘、肠闭锁或狭窄、肛门直肠畸形等病鉴别。用小指肛诊后,巨结肠病儿能大量排出胎便及气体,随之症状缓解。因此,对新生儿肠梗阻,常规做肛诊检查,不仅有助于诊断和治疗新生儿巨结肠,而且可避免或减少错误诊治。 6个月以上病儿,有慢性便秘等症状、又有新生儿巨结肠史,诊断先天性巨结肠较容易,否则须除外肛门直肠畸形等病所致续发性巨结肠,以及克汀病、饮食性便秘、特发性巨结肠等症。肛诊时,短段型先天性巨结肠的肛门紧缩,直肠明显扩张积便或积气;普通型先天性...[详细]
1.内科治疗?对轻型的先天性巨结肠患儿或有全身感染症状,手术无法耐受者可用非手术疗法维持营养和发育。用缓泻剂或定时用生理盐水灌肠,以避免粪便淤积。 2.结肠造瘘术?当患儿发生急性肠梗阻,或有肠穿孔、腹膜炎趋向,或伴有小肠结肠炎,或是全结肠无神经节症,应行结肠造瘘术。造口部位应尽量选择靠近扩张肠管作单口造瘘。 3.手术治疗?新生儿期患儿应尽量保守治疗,待1岁左右再做根治手术。成人期患者症状加重,经保守治疗无效者亦应行根治术。根治手术要求切除距肛门齿状线1~2cm处以上的狭窄直肠及狭窄段上5cm以上的扩张结肠。常用的术式有下面三种: (1)拖出型直肠、乙状结肠切除术(Swensen手术)。 (2)结肠切除、直肠后结肠拖出术(Duhamel手术)。 (3)直肠黏膜剥离、结肠于直肠肌层内拖出切除术(Soave手术)。[收起]
1.内科治疗?对轻型的先天性巨结肠患儿或有全身感染症状,手术无法耐受者可用非手术疗法维持营养和发育。用缓泻剂或定时用生理盐水灌肠,以避免粪便淤积。 2.结肠造瘘术?当患儿发生急性肠梗阻,或有肠穿孔、腹膜炎趋向,或伴有小肠结肠炎,或是全结肠无神经节症,应行结肠造瘘术。造口部位应尽量选择靠近扩张肠管作单口造瘘。 3.手术治疗?新生儿期患儿应尽量保守治疗,待1岁左右再做根治手术。成人期患者症状加重,经保守治疗无效者亦应行根治术。根治手术要求切除距肛门齿状线1~2cm处以上的狭窄直肠及狭窄段上5cm以上的扩张结肠。常用的术式有下面三种: (1)拖出型直肠、乙状结肠切除术(...[详细]
新生儿HD诊断治疗均十分困难,多数文献报道,采用常规洗肠等保守疗法,半年内病死率为50%~70%,1年达70%~90%。肠炎发生率为20%~30%左右,肠穿孔约为3.4%~6.4%。国内余氏亦报道新生儿保守治疗及肠造瘘术后总死亡率仍高达40%。新生儿根治手术死亡率为3.1%~12%,近年来也有少数病例报道根治术未发生死亡者。因此对新生儿的HD诊治应特别慎重,根据患儿一般情况及病变肠管的长度、医院设备及条件,可分别选择中西医结合非手术疗法,经肛门路手术及根治手术。 虽然婴幼儿HD随着年龄的增长,其手术危险性逐渐降低。但是根据国外大宗病例报道,根治术后并发症仍然较多。术后伤口感染约占10%,吻合口漏约7.2%,肠梗阻11.2%。远期随访肛门失禁仍有13.6%,便秘复发9.4%,肠炎7%,死亡2.2%,需再次手术者占8.1%~12.9%。国内随访1017例,大致和上述并发症相同。近年来Shonc(1994)报道术后早期并发症占25%以上,晚期并发症近40%,Boley术后约一半出现早期梗阻症状,78%发生肠炎,术后便秘复发占21.9%,肠瘘3.3%,死亡3.3%。Skaba报道94例Kasai术后,伤口感染12.7%,吻合口裂开11.7%,吻合口狭窄10.6%。该学者收集文献共4431例,术后死亡率为0%~3.4%,吻合口狭窄3%~21%,吻合口漏3.4%~13.3%,术后便秘复发约占10%左右,肠炎5%~10%。从上述资料已不难看出,HD根治手术后并发症仍然多而严重,尤其是远期随访时仍有10%~15%需再手术;晚期病死率约2.2%~3.4%。[收起]
新生儿HD诊断治疗均十分困难,多数文献报道,采用常规洗肠等保守疗法,半年内病死率为50%~70%,1年达70%~90%。肠炎发生率为20%~30%左右,肠穿孔约为3.4%~6.4%。国内余氏亦报道新生儿保守治疗及肠造瘘术后总死亡率仍高达40%。新生儿根治手术死亡率为3.1%~12%,近年来也有少数病例报道根治术未发生死亡者。因此对新生儿的HD诊治应特别慎重,根据患儿一般情况及病变肠管的长度、医院设备及条件,可分别选择中西医结合非手术疗法,经肛门路手术及根治手术。 虽然婴幼儿HD随着年龄的增长,其手术危险性逐渐降低。但是根据国外大宗病例报道,根治术后并发症仍然较多。术后伤口感染约占10...[详细]


