-
科室:
内分泌科
-
别名:
特发性促性腺激素不足性性腺机能减退
特发性低促性腺素功能减退症
-
症状:
性发育慢
-
发病部位:
暂无
-
多发人群:
所有群体
-
相关疾病:
暂无
低促性腺激素性性腺功能减退(HH)包括一大组疾病,先天性GnRH神经元缺陷、垂体促性腺激素缺乏或分子结构异常、慢性全身性疾病、精神应激、严重体重丢失或长期剧烈运动都可引起促性腺激素缺乏。此外,促性腺激素缺乏也是一些先天性综合征(如Prader-Willi综合征,Laurence-Moon-Biedl综合征等)的一个组成成分。促性腺激素缺乏的程度也具有不均一性,一个极端是完全无青春期性成熟,另一个极端是青春期延迟,中间是不同程度的过度类型。
IHH的遗传特性在家系分析资料中不是单一类型,至少有3种不同的遗传方式。 一些家系分析的结果发现一个嗅觉缺失的父亲生育了嗅觉缺失和(或)性腺功能减退的儿子,而所生的女儿性腺发育和嗅觉正常。更为有趣的例子是父亲是卡尔曼综合征患者,经过长期人绒毛膜促性腺激素治疗后,结婚并生育了患卡尔曼综合征的儿子。这些家系例证与常染色体显性遗传一致。另一些家系则是祖代和父代家庭成员没有发现异常,第3代的儿女中男性和女性都有嗅觉缺失和性腺功能减退患者,这种遗传方式显然符合常染色体隐性遗传。此外,还有一些家系父亲正常,母亲是携带者,生育的子女中,只有男性出现性腺功能减退和(或)嗅觉缺失。而女儿结婚后,生育的女性子代表现正常,而男性子代是卡尔曼综合征患者,属于X-连锁遗传。这种遗传的不均一性不仅表现在遗传方式上,即使是同一遗传方式也存在表达的不均一性,即同一家系发病的成员中,可有单纯性腺功能减退而无嗅觉缺失,或只有嗅觉缺失而无性腺功能减退;嗅觉缺失的程度也存在差异,一些受累家庭成员的嗅觉缺失是不完全的,只有嗅觉减退。一个更为突出的例证是一对20岁同卵孪生兄弟,其中一个是典型的卡尔曼综合征患者,而另1个只有嗅觉缺失,生殖器官发育正常,血浆促性腺激素和睾酮水平正常。[收起]
IHH的遗传特性在家系分析资料中不是单一类型,至少有3种不同的遗传方式。 一些家系分析的结果发现一个嗅觉缺失的父亲生育了嗅觉缺失和(或)性腺功能减退的儿子,而所生的女儿性腺发育和嗅觉正常。更为有趣的例子是父亲是卡尔曼综合征患者,经过长期人绒毛膜促性腺激素治疗后,结婚并生育了患卡尔曼综合征的儿子。这些家系例证与常染色体显性遗传一致。另一些家系则是祖代和父代家庭成员没有发现异常,第3代的儿女中男性和女性都有嗅觉缺失和性腺功能减退患者,这种遗传方式显然符合常染色体隐性遗传。此外,还有一些家系父亲正常,母亲是携带者,生育的子女中,只有男性出现性腺功能减退和(或)嗅觉缺失。而女儿结婚后,生育的...[详细]
X-连锁遗传型卡尔曼综合征的分子遗传学基础已经确定,X染色体短臂的末端部分是假常染色体区,这个区段的DNA序列与Y染色体的假常染色体区同源。当减数分裂时,X和Y染色体的这个区段会发生DNA配对和交换。假常染色体区的基因在男女两性都是双倍剂量,因而能避免X染色体的失活。这个区段含有PHOX/SHOX(矮身材)基因、MI C2(一种细胞表面抗原)基因、点状软骨发育不全基因、智能减退基因、STS(类固醇硫酸酯酶)基因和KAL1(卡尔曼综合征)基因等。用基因图技术可以确定KAL1基因位于Xp22.3区,靠近STS基因。在X-连锁型卡尔曼综合征患者中,已发现KAL1基因存在大的或小的缺失,点突变和各种无义突变,导致构架改变和过早出现终止密码子。少数患者在密码区未发现有突变,变异的部位可能是在启动子区。邻近基因的连带缺失可引起卡尔曼综合征、X-连锁的鱼鳞癣(STS基因缺失)、智能减退和(或)点状软骨发育不全。KAL1基因的不同突变方式转录出不同的基因产物,后者与临床表现的不均一性有关。现在已可应用Southern印迹技术分析胎儿的DNA在产前诊断X-连锁型卡尔曼综合征。KAL1基因长约1.5Mb,编码1个680氨基酸的糖蛋白,在功能上这个蛋白具有细胞外神经黏附分子的特性,可能是GnRH神经元从胎儿时期的嗅板迁徙到下丘脑内侧底部的引路蛋白。关于基因治疗目前尚无可行的方案,但是KAL1基因及其编码蛋白的结构已经阐明,有朝一日通过基因治疗补充正常结构蛋白以预防卡尔曼综合征不是完全不可能的事情。至于常染色体显性遗传和隐性遗传2种类型的致病基因现在仍所知甚少,是否在某条常染色体中存在着和KAL1相似的基因,还是KAL1基因亦与常染色体遗传类型有关?此外,单纯表达低促性腺激素性性腺功能减退而无嗅觉减退的患者是否亦是KAL1基因起着关键的作用?这些问题还有待于进一步的研究结果来回答。 GnRH受体(GnRH-R)基因已经克隆出来,定位于第4号染色体长臂,是一种G蛋白耦联膜受体,有7个穿膜区,N-端在细胞外,但是没有细胞内C-端。受体的激活使磷酸酯酶活性增高和促进G蛋白介导的细胞内钙动员。最近已有一个家系因GnRH-R突变引起IHH的病例报道,患者男性,22岁,嗅觉正常,未发现存在其他畸形。无胡须,阴毛Tanner Ⅲ期,阴茎长6cm,睾丸容积8ml。精液分析精子密度3.91×106/ml,43%形态正常,5%活动,果糖和枸橼酸盐浓度显著低于正常。血清睾酮水平2.8nmol/L,LH4.OU/L,FSH5.9U/L。LH和FSH对GnRH(100μg)兴奋试验的反应正常。LH8h脉冲分析(每10min采血1次)提示脉冲频率正常,脉冲幅度减低(只相当于正常脉冲幅度的1/5)。患者的大姐14岁时青春期启动,原发闭经和不育,B超发现双侧卵巢小,未见优势卵泡。患者的父母和二姐性发育正常。GnRH-R基因3个外显子DNA的扩增产物测序显示患者和他的大姐有两处复合杂合子突变,一处是受体细胞外第1环存在Gln106Arg突变,形成核苷酸317的腺嘌呤被鸟嘌呤取代;第二处是细胞内第3环的Arg262Gln突变,使785位核苷酸的鸟嘌呤被腺嘌呤替代。其母亲只携带Gln106Arg突变,父亲和患者的二姐只携带Arg262Gln突变。GnRH-R的细胞外第1环与受体的结合能力有关,实验性Asn102Ala突变使GnRH-R完全失去了与GnRH结合的能力,Gln106Arg突变仍保存了一部分生物学反应,可能是受体激素复合物相对不稳定所致。GnRH-R细胞内第3环是受体信号传递的关键区域,Arg262G1n突变不影响受体与激素的结合,而影响G蛋白的耦联和受体内在化等受体后反应。 GnRH基因缺失在小鼠的实验研究中已成功地证明会发生低促性腺激素性性腺功能减退,提示GnRH基因突变是IHH的致病原因之一。但是对少数IHH患者的GnRH基因测序尚未发现存在缺失或点突变等异常。 为了研究和了解下丘脑GnRH的分泌方式和特性,一般是采取下列2种方法:一是对正常人频繁采集外周血测定LH和(或)FSH,分析其脉冲频率和幅度。根据前述每个GnRH脉冲能诱发出1个LH(和FSH)分泌脉冲的原则,LH的脉冲频率必然是GnRH脉冲频率的反映。LH脉冲幅度的高低则是每个GnRH脉冲的释放量和性激素反馈调节共同作用的结果。二是研究IHH患者或动物模型的GnRH分泌方式,这对了解GnRH脉冲分泌的机制和制定外源性GnRH替代治疗方案是非常重要的。在进行脉冲分析时,应该认识到:①虽然每个LH和FSH脉冲都是GnRH脉冲的反映,但是,不是每一个GnRH脉冲都会被垂体转录为可识别的LH和FSH脉冲,即LH和FSH脉冲数目不一定与GnRH的脉冲数目完全相等;②所有的LH和FSH脉冲应该是可以观察到的,如果遗失一部分资料,对脉冲频率和幅度的分析结论可能不准确;③影响脉冲分析的因素有激素测定方法的灵敏度、确定脉冲的方法和采血密度等,其中影响最大的是采血密度,采血密度与脉冲间期的长短密切相关,最适的采血密度是5~10min 1次。 以目前常用的放免测定方法,LH的脉冲频率与GnRH脉冲的一致率比FSH脉冲高,这是因为FSH的半衰期较长,LH和FSH固有的分泌方式不同以及性激素、抑制素、激动素和卵泡抑制蛋白对2种促性腺激素的调节作用不同所致。例如成年男子的睾酮水平或女子卵泡中期的E2水平对FSH的抑制作用大于LH,以及GnRH脉冲频率的改变可以改变LH/FSH释放的比例等。正常成年男子的LH脉冲间期约为90~120min,即24h出现12~16个脉冲。正常成年男子的LH脉冲频率有相当的变异,24h只有7个脉冲仍有正常的性发育和生育能力的例子已有报道。正常成年女子的LH脉冲间期有明显的月经周期影响,卵泡早期(月经第2~6天)约为100min,脉冲幅度中等,睡眠时脉冲释放几乎完全停止。卵泡中期(月经第7~10天)约为60min,脉冲幅度降低,睡眠时有脉冲出现。卵泡晚期(月经第11~14天)约为70min,脉冲幅度增加,昼夜脉冲无差别。黄体早期(排卵第1~4天)约为100min,大脉冲(幅度>15U/L)和小脉冲(<5U/L)并存。黄体中期(排卵第5~9d)约为200min,小脉冲占50%。黄体后期(排卵第10~14天)约为300min,大脉冲减少到1~2个/24h,几乎全部为小脉冲。 男性IHH患者的LH脉冲分泌异常(至少每10分钟采血1次)有下列几种方式:①无脉冲分泌,和青春期前儿童的情况一样。这种分泌方式在IHH男性患者中最多见,约占全部病例的75%。②夜间出现脉冲分泌,和青春期启动早期儿童的情况相似。这些患者往往有青春期启动史,睾丸相对较大,但是以后出现停滞,未能完成青春期发育过程,因而又称为青春期停滞型。③脉冲幅度低,这种小脉冲不足以兴奋睾丸的赖迪细胞合成和分泌睾酮。④脉冲频率不足,24h不足7个脉冲,在脉冲出现时睾酮的分泌可达21.0mmol/L,但是随着LH脉冲的消失,血清睾酮水平逐渐降低,不能维持在正常范围,不能维持生殖器官和第二性征的发育。见图1。女性IHH或下丘脑闭经患者的LH脉冲分泌异常和在男性IHH患者所见到的相同,亦可分为无脉冲型、青春期停滞型、脉冲幅度减低型和脉冲频率减慢型4种分泌方式异常。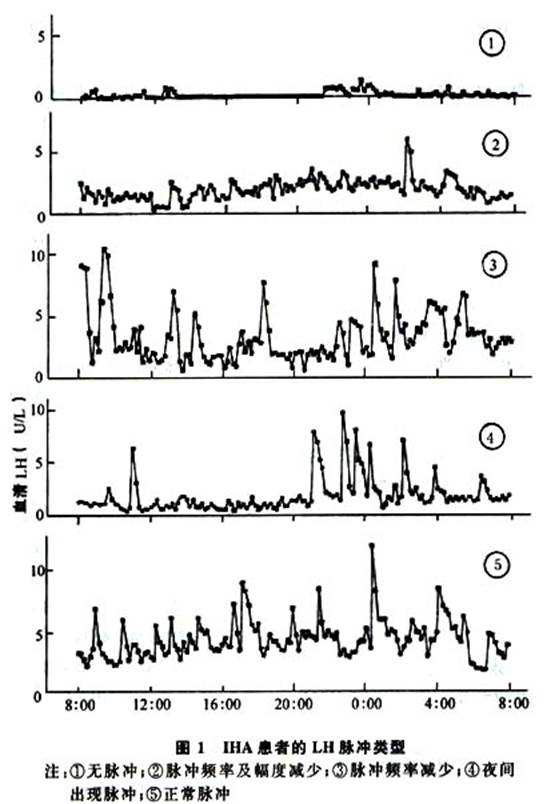 [收起]
X-连锁遗传型卡尔曼综合征的分子遗传学基础已经确定,X染色体短臂的末端部分是假常染色体区,这个区段的DNA序列与Y染色体的假常染色体区同源。当减数分裂时,X和Y染色体的这个区段会发生DNA配对和交换。假常染色体区的基因在男女两性都是双倍剂量,因而能避免X染色体的失活。这个区段含有PHOX/SHOX(矮身材)基因、MI C2(一种细胞表面抗原)基因、点状软骨发育不全基因、智能减退基因、STS(类固醇硫酸酯酶)基因和KAL1(卡尔曼综合征)基因等。用基因图技术可以确定KAL1基因位于Xp22.3区,靠近STS基因。在X-连锁型卡尔曼综合征患者中,已发现KAL1基因存在大的或小的缺失,点突变和各种无...[详细]
[收起]
X-连锁遗传型卡尔曼综合征的分子遗传学基础已经确定,X染色体短臂的末端部分是假常染色体区,这个区段的DNA序列与Y染色体的假常染色体区同源。当减数分裂时,X和Y染色体的这个区段会发生DNA配对和交换。假常染色体区的基因在男女两性都是双倍剂量,因而能避免X染色体的失活。这个区段含有PHOX/SHOX(矮身材)基因、MI C2(一种细胞表面抗原)基因、点状软骨发育不全基因、智能减退基因、STS(类固醇硫酸酯酶)基因和KAL1(卡尔曼综合征)基因等。用基因图技术可以确定KAL1基因位于Xp22.3区,靠近STS基因。在X-连锁型卡尔曼综合征患者中,已发现KAL1基因存在大的或小的缺失,点突变和各种无...[详细]
IHH患者在青春期前如无小阴茎、隐睾或其他器官或躯体异常,一般不易发现。大多数患者是因为到了青春期年龄无性发育而求医,少数患者有过青春期启动,但是中途发生停滞,性成熟过程未能如期完成,这些患者的睾丸容积较大,可达到青春期Ⅱ期或Ⅲ期的水平。约90%的患者的喉结小,阴毛和腋毛缺如,少数患者可有少量阴毛生长(Tanner阴毛Ⅱ期)。80%的患者骨龄落后于实际年龄。40%有嗅觉缺失或嗅觉减退。20%有男子乳腺增生。可有小阴茎、隐睾和输精管缺如。还可伴发其他躯体或器官异常,如面颅中线畸形:兔唇、腭裂、腭弓高尖和舌系带短。神经系统异常:神经性耳聋、眼球运动或视力异常、红绿色盲、小脑共济失调、手足连带运动和癫痫。肌肉骨骼系统异常:骨质疏松、肋骨融合、第4掌骨短、指骨过长和弓形足。其他系统异常:皮肤牛奶咖啡斑。肾发育不全或畸形、先天性心血管病(主动脉弓右位、锁骨下动脉狭窄、房室传导阻滞和右心肥大等)。身高一般正常,少数患者身材矮。肥胖,智力一般正常。 IHH的临床表现与下丘脑GnRH脉冲分泌异常的类型有关,将男性IHH患者的临床表现和LH脉冲分析的结果对比观察可以发现,无脉冲分泌型患者可分为两部分:第一部分患者例数最多,病情较重,从未有过自发的青春期发育,睾丸小,平均容积约为3ml,睾丸活检组织学所见与青春期前儿童毫无二致,常常伴有嗅觉缺失或嗅觉减退。平均血清LH水平为1.7±0.3 U/L,FSH 2.1±0.2 U/L。少数患者的LH和FSH减低到可检出范围以下。可有隐睾和阴茎小。第二部分患者例数较少,约占本类型患者的23%。病情较轻,有过不完全的自发性青春期发育,中间发生了停滞。睾丸较大,容积可达3~8ml,睾丸活检显示无精子发生、成熟停滞或甚至正常的精子发生。嗅觉一般正常,无隐睾和小阴茎。但是,血清LH和FSH的平均水平与第一部分患者无明显差别。青春期停滞型患者夜间有LH脉冲分泌,在14~15岁时有过青春期启动,有一定程度的性成熟表现,睾丸容积可达8~12ml,可有自发的阴茎勃起和性冲动,但是性发育未能继续进行下去,停留在青春期早期阶段。无嗅觉缺失或减退,血清LH和FSH水平平均可达正常值范围低限。脉冲幅度减低型患者睾丸容积3~6ml,血清睾酮水平1.1~3.5nmol/L,LH和FSH水平低于正常。脉冲频率减慢型患者由于在脉冲出现时有睾酮分泌,血清睾酮水平波动在3.8~21.0mmol/L之间,有一定程度的性发育,睾丸容积可达10~15ml,无嗅觉缺失,可有男子乳腺增生,是IHH中病情最轻的一种类型。[收起]
IHH患者在青春期前如无小阴茎、隐睾或其他器官或躯体异常,一般不易发现。大多数患者是因为到了青春期年龄无性发育而求医,少数患者有过青春期启动,但是中途发生停滞,性成熟过程未能如期完成,这些患者的睾丸容积较大,可达到青春期Ⅱ期或Ⅲ期的水平。约90%的患者的喉结小,阴毛和腋毛缺如,少数患者可有少量阴毛生长(Tanner阴毛Ⅱ期)。80%的患者骨龄落后于实际年龄。40%有嗅觉缺失或嗅觉减退。20%有男子乳腺增生。可有小阴茎、隐睾和输精管缺如。还可伴发其他躯体或器官异常,如面颅中线畸形:兔唇、腭裂、腭弓高尖和舌系带短。神经系统异常:神经性耳聋、眼球运动或视力异常、红绿色盲、小脑共济失调、手足连带运动和...[详细]
1.血清性激素水平低于正常,LH和FSH水平正常低限或低于正常。 2.GnRH兴奋试验无论是男性或女性患者,LH的分泌反应一般是减低的,少数患者完全无反应或反应正常。同一患者的LH反应可以和FSH反应不一致。 3.血清PRL基础水平正常,PRL对促甲状腺激素释放激素(TRH)和氯丙嗪兴奋试验的反应一般正常,少数反应减低,个别反应过强。 4.患者的甲状腺功能(临床表现和TT4,TT3,FT4,FT3和TSH)正常,TRH兴奋TSH试验一般反应正常,ACTH和皮质醇的昼夜节律正常,皮质醇对ACTH兴奋的反应正常。 5.尿浓缩功能正常。 以上资料说明除下丘脑-垂体-性腺轴系外,腺垂体的PRL,GH,ACTH和TSH功能正常,神经垂体功能也正常。[收起]
1.血清性激素水平低于正常,LH和FSH水平正常低限或低于正常。 2.GnRH兴奋试验无论是男性或女性患者,LH的分泌反应一般是减低的,少数患者完全无反应或反应正常。同一患者的LH反应可以和FSH反应不一致。 3.血清PRL基础水平正常,PRL对促甲状腺激素释放激素(TRH)和氯丙嗪兴奋试验的反应一般正常,少数反应减低,个别反应过强。 4.患者的甲状腺功能(临床表现和TT4,TT3,FT4,FT3和TSH)正常,TRH兴奋TSH试验一般反应正常,ACTH和皮质醇的昼夜节律正常,皮质醇对ACTH兴奋的反应正常。 5.尿浓缩功能正常。 以...[详细]
1.有阳性家族史的患者应尽可能进行常染色体检查,做家系分析。 2.检查嗅觉、红、绿色盲等。
IHH的诊断有相当大的难度,对可疑的IHH患者要详细采集病史,了解宫内和幼年生长发育的情况,是否存在生长停滞。一般地说,IHH的生长停滞相对较轻,身高基本上在同龄儿童正常身高的范围以内,但是,由于性激素长期处于低水平,过了青春期年龄的患者四肢生长过度,形成类无睾体型(下部量>上部量,指距>身高)。有阳性家族史的患者应尽可能进行家系分析,目前已证实的遗传方式有3种:X-连锁、常染色隐性和常染色体显性,存在其他遗传方式的可能性不能排除。体格检查要特别检查嗅觉,有嗅觉缺失或嗅觉减退者病情较严重。一部分患者可能有红、绿色盲,腭裂和(或)兔唇等中线发育畸形,其他发育异常可见于中枢神经系统、骨骼和肾脏等,智力一般正常。儿童期阴茎小,睾丸可能下降不全。青春期年龄后没有性发育的征象。骨龄落后于实际年龄。肾上腺皮质功能初现在6~8岁时如期启动。血清促性腺激素和性激素仍处于青春期前的低水平,GH水平正常。当根据临床表现和实验检查仍不足以确定诊断时,则需要长期的追随观察,一般把18岁作为一个界限,超过18岁仍无青春期启动者可诊断为IHH。[收起]
IHH的诊断有相当大的难度,对可疑的IHH患者要详细采集病史,了解宫内和幼年生长发育的情况,是否存在生长停滞。一般地说,IHH的生长停滞相对较轻,身高基本上在同龄儿童正常身高的范围以内,但是,由于性激素长期处于低水平,过了青春期年龄的患者四肢生长过度,形成类无睾体型(下部量>上部量,指距>身高)。有阳性家族史的患者应尽可能进行家系分析,目前已证实的遗传方式有3种:X-连锁、常染色隐性和常染色体显性,存在其他遗传方式的可能性不能排除。体格检查要特别检查嗅觉,有嗅觉缺失或嗅觉减退者病情较严重。一部分患者可能有红、绿色盲,腭裂和(或)兔唇等中线发育畸形,其他发育异常可见于中枢神经系统、骨骼和肾脏等,...[详细]
1.性激素替代治疗 性激素替代治疗是IHH的基本治疗措施。性激素替代治疗的目的是促进第二性征的发育和维持性功能。替代治疗的原则是模拟正常的青春期过程,正常的青春期一般历时4~5年。因此,替代治疗的性激素剂量要从小量开始,以避免骨骺过早闭合导致矮身材。约在1年后增量至成人常规剂量,持续3~4年。 (1)睾酮替代治疗:男孩的睾酮替代治疗一般在14岁开始,可供选择的睾酮制剂包括口服剂、肌内注射剂和皮肤贴剂。 十一酸睾酮(TU):化学结构为3-氧雄烷-4烯-17β-+一酸,有较强的嗜脂性。口服剂在口服后和脂肪微粒一起在小肠经淋巴系统吸收,然后进入体循环,避免了其他口服睾酮制剂在肝脏被灭活(首过效应)和肝毒性的缺点。口服后血浆达峰时间有明显的个体差异,平均约为4h,在体内酯键被降解而释放出睾酮。连续服用后,血浆睾酮水平逐渐升高,在2~3周后达到平高,并长期保持稳定。十一酸睾酮注射剂单剂肌内注射后血浆睾酮达峰时间约在第7天,第21天后恢复到注射前水平。替代治疗开始剂量为口服剂40mg/d,肌注剂每4周50~100mg,9~12个月后逐渐增加剂量至120~160mg/d,分2次口服或200mg,每2~3周肌注1次。持续3~4年。此后可适当减少剂量,以维持第二性征和性功能达到或接近正常为原则。 庚酸睾酮(testoserone enanthate):化学结构为3-氧雄烷-4烯-17β-庚酸,只有肌注剂,肌注后的吸收、分布和降解代谢和十一酸睾酮注射剂相似。替代治疗剂量开始每4周肌注50~100mg,9~12个月后增量到200mg,每2~3周肌注1次,连续3~4年。 睾酮皮肤贴剂:每贴每天释出睾酮4~6mg,每天一贴,贴于阴囊或其他部位皮肤,可以获得比肌注剂更符合正常生理的血浆睾酮浓度,但费用较高。 睾酮长期替代治疗可以产生的不良反应包括痤疮或原有的痤疮加重、膀胱刺激症状(尿频)、男子乳腺增生、乳腺痛、性欲亢进、痛性勃起(与剂量过大有关)、非特异性附睾炎(阴囊或腹股沟部疼痛、寒战)、下肢水肿、体重增加、红细胞增多(头晕、腹痛、腹泻、便秘)、过敏反应(皮疹、哮喘、神经血管性水肿)、注射局部反应(瘙痒、发红、疼痛)、肝功能异常、免疫力减退(继发真菌感染或其他细菌感染)、脂质代谢紊乱和精神障碍(欣快感、注意力不集中、烦躁、情绪不稳定、失眠和暴力倾向)。 (2)雌激素替代治疗:女孩的雌激素替代治疗在13岁开始,目的是获得接近正常的第二性征发育和撤退出血。常用雌激素为炔雌醇(乙炔雌二醇)和结合雌激素,炔雌醇(乙炔雌二醇)的化学结构为19去甲孕-1,3,5-三烯-20炔-3,17α-二醇,是一种强效的口服雌激素,其活性为雌二醇的8倍,己烯雌酚的20倍。口服后达峰时间约为2h,t1/2为6~14h,生物利用度50%。结合雌激素是从妊娠马尿中提取出来的复合成分雌激素,主要成分是雌酮、马烯雌酮和17α-二氢马烯雌酮,其他含量较少的成分还有17α-雌二醇、马萘雌酮和17α-二氢马萘雌酮等。开始剂量[收起]
1.性激素替代治疗 性激素替代治疗是IHH的基本治疗措施。性激素替代治疗的目的是促进第二性征的发育和维持性功能。替代治疗的原则是模拟正常的青春期过程,正常的青春期一般历时4~5年。因此,替代治疗的性激素剂量要从小量开始,以避免骨骺过早闭合导致矮身材。约在1年后增量至成人常规剂量,持续3~4年。 (1)睾酮替代治疗:男孩的睾酮替代治疗一般在14岁开始,可供选择的睾酮制剂包括口服剂、肌内注射剂和皮肤贴剂。 十一酸睾酮(TU):化学结构为3-氧雄烷-4烯-17β-+一酸,有较强的嗜脂性。口服剂在口服后和脂肪微粒一起在小肠经淋巴系统吸收,然后进入体循环,避免了其他口服睾酮制剂在肝...[详细]
1.长期静脉给药治疗会给患者生活带来不便,降低了患者对治疗的顺应性。有发生静脉炎的危险。 2.雌激素替代治疗已有报道,长期大剂量服用雌激素使乳腺癌的发病率增高。子宫内膜癌的发病率增加4倍,在停药后10年内仍有这种危险。 3.心肌梗死、肺梗死和血栓性静脉炎的发病率增加。此外可有血压增高、体重增加、水肿、高血钙、糖耐量减低和加重卟啉病等。
1.在雌激素替代治疗期间应定期做乳腺,妇科,心血管系统等方面的检查。 2.性激素替代治疗剂量要从小量开始,以避免骨骺过早闭合导致矮身材。


