概述
本病是一种亚急性的家族性TSE,临床上以治疗无效的顽固失眠、自主神经机能失调和运动体征为特征,组织病理学以丘脑前腹侧和背内侧神经核选择性萎缩为特征。早先,该病与家族性CJD混同。1986年,意大利Bologna大学医学院Lugaresi等首先报道并详细描述了本病的第一个病例,命名为致死性家族性失眠症。从此,人朊病毒病增加了一个新成员。
病因
致死性家族性失眠病(fatal familial insomnia,FFI)是最近被确认的家族性人类prion疾病。 PrP基因突变 FFI是常染色体显性遗传性疾病,与129Met 178Asn单元型相关联。已证实无亲缘关系的5个家族的17例FFI病人有PRNP 178密码子突变,该密码子的单一等位基因的Asp被Asn替代(178Asn)。另外,FFI病人PRNP基因129密码子Met/Val多态性也明显分布不匀。这种多态性在正常白种人群体中的分布是Met/Met 0.37、Met/Va10.51、Va1/Va10.12;而在FFI患者中则是Me/Met 0.82、Met/...[详细]
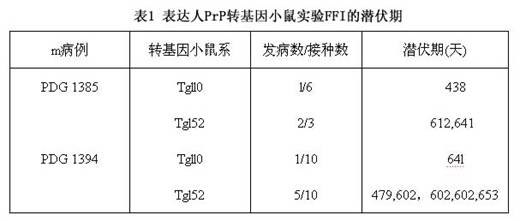 FFI患者脑内PrPsc的水平低 FFI患者的脑匀浆中含PrPsc,但水平比CJD等朊病毒病低。Medori等检查5例,4例阳性。以蛋白酶K处理后作Westen印迹产生29和27kDa 2个主要片断,而散发性CJD则恒产生29、25和21kDa 3个主要片断。Monar等报告,FFI产生28和26kDa 2个主要片断,底片长时间曝光后另可见一19kDa的次要片断。FFI PrPsc的模式是否也和其他家族性朊病毒病不同、即为FFI所独有,尚有待研究确定。其抗蛋白酶PrP的量似乎与病程相关,而和各脑区病损的严重程度无关;因为它在FFI病人的丘脑中的量并不显著高于其他脑区,而在1例丘脑无组织学病损患者的基底神经节内却大量存在。
FFI患者脑内PrPsc的水平低 FFI患者的脑匀浆中含PrPsc,但水平比CJD等朊病毒病低。Medori等检查5例,4例阳性。以蛋白酶K处理后作Westen印迹产生29和27kDa 2个主要片断,而散发性CJD则恒产生29、25和21kDa 3个主要片断。Monar等报告,FFI产生28和26kDa 2个主要片断,底片长时间曝光后另可见一19kDa的次要片断。FFI PrPsc的模式是否也和其他家族性朊病毒病不同、即为FFI所独有,尚有待研究确定。其抗蛋白酶PrP的量似乎与病程相关,而和各脑区病损的严重程度无关;因为它在FFI病人的丘脑中的量并不显著高于其他脑区,而在1例丘脑无组织学病损患者的基底神经节内却大量存在。发病机制
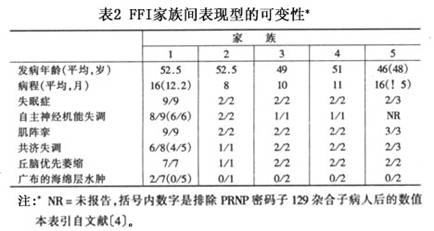
本病的组织病理学以丘脑萎缩为主(表2)。丘脑前腹侧和背内侧核恒严重受侵,丘脑中央内侧核和枕核也经常受害,其他丘脑神经核是否受累则变化不定。皮质通常显示极轻度至中度的星形神经胶质增生,且累及深层,并延伸至浅在的白质。被检病例中。只两例见广布的海绵层水肿,这两例病程较长(2532个月),也具有周期性的EEG;一例病程13周的病人灶性海绵层水肿局限于海马回下脚。下橄榄体常严重萎缩,小脑皮质则仅呈极轻度至中度的萎缩。其他脑区一般正常。受检的14例中,4例基底神经节有极轻度的神经胶质增生,1例上丘有中度神经胶质增生。...[详细]
临床表现
一般的说,病初有三种不同的表现:①睡眠障碍,通常病人自诉失眠、睡眠期间激动、多梦;②运动体征,如构音障碍和共济失调;③记忆障碍。随着疾病的发展,病人呈现FFI的全部症状,涉及运动、内分泌和自主神经等系统。具体如下: 1.睡眠和失眠 进行性失眠,失眠日益增重,这已在接受多导睡眠描记法(polysomnography)或通宵EEG记录检查的4个家族的8例病人得到证明。用安定和巴比妥酸盐等安眠药治疗无效。病人还呈现进行性的似梦中状态和幻觉,病末期呈木僵和昏睡状态。 2.识别机能 劳动记忆,注意力和视运动(visumotor)功能受损,但仍保持球智力(global inte...[详细]
并发症
并发脑萎缩。
实验室检查
外周血常规检查正常
其他辅助检查
1.与深睡眠相关的EEG活动减少或消失。 2.[18F]PET检查,丘脑区代谢优先减低。 3.EEG 睡眠期间:δ-活动、睡眠梭形波、K复合体减少,甚至完全消失;快速眼运动(REM)相异常;觉醒期间:进行性扁平的背景活动,缓慢的活动,不能产生药物诱导的睡眠活动[受检的7例病人只2例见周期性峰活动,这两例病人病程长,分别为25和32个月。脑皮质广布海绵状层水肿(spongiosis)]。 4.阳电子反射断层摄影术(PET)扫描 检4例,2例代谢减退(-36%)、实际上局限于丘脑前部;另2例代谢减退也存在于脑皮质(特别是额叶和顶叶)(-40%)、海马、基底神经节和...[详细]
诊断
1993年Gambetti等提出了如下的诊断标准: 1.常染色显体性疾病,成年期发病,病程6~32个月。 2.呈现治疗无效的顽固失眠症、家族性自主神经机能异常、记忆障碍、共济失调和(或)肌阵挛、锥体束和锥体外束征。 3.129Met和178Asn单元型。 具备标准15中任何2项均可作FFI的可疑诊断。标准6兼有其他任何一项标准即可确诊FFI。
具备标准15中任何2项均可作FFI的可疑诊断。标准6兼有其他任何一项标准即可确诊FFI。
具备标准15中任何2项均可作FFI的可疑诊断。标准6兼有其他任何一项标准即可确诊FFI。
治疗
对症及支持治疗可减轻症状,改善生活质量,但至今尚无有效的病原治疗。有报道认为刚果红、二甲基亚砜、酚噻嗪、氯丙嗪、分支多胺、磷脂酶C、抗朊毒体抗体及寡肽等可能对延缓病情有一定作用,但效果及适用性有待证实。
预后
预后极差,已知病例无一例外均告死亡。


