-
科室:
暂无
-
别名:
phenylalanine hydroxylase deficiency of children
小儿苯丙氨酸羟化酶缺乏症
小儿苯丙酮酸尿
-
症状:
智力低下
生长缓慢
-
发病部位:
暂无
-
多发人群:
儿童
-
相关疾病:
染色体异常
苯丙酮尿症(phenylketonuria,PKU)是由于苯丙氨酸代谢途径中,酶缺陷所导致的较为常见的一种常染色体隐性遗传性疾病,在1934年因Foiling最早发现病人尿中含有大量的苯丙酮酸而得名。1947年Jervis对病人进行苯丙氨酸负荷实验,揭示PKU发病的生化基础是肝脏苯丙氨酸代谢障碍。1953年,德国的Bickel首先报道用低苯丙氨酸奶方治疗PKU病人获得成功。1963年Guthrie开展了PKU新生儿筛查。1983年Woo克隆了PKU的致病基因苯丙氨酸羟化酶基因,为基因诊断和产前诊断开辟了道路。
本病属常染色体隐性遗传性疾病,系苯丙氨酸代谢途径中酶的缺陷所致。
苯丙氨酸(phenylalanine,Phe)是人体必需氨基酸,食入体内的Phe一部分用于蛋白质的合成,一部分通过苯丙氨酸羟化酶作用转变为酪氨酸,仅有少量的Phe经过次要的代谢途径在转氨酶的作用下转变成苯丙酮酸。 PKU是因苯丙氨酸羟化酶(phenylalanine hydroxylase,PAH)基因突变导致PAH活性降低或丧失,Phe在肝脏中代谢紊乱所致。PKU患者苯丙氨酸羟化酶缺乏,酪氨酸及正常代谢产物减少,血Phe含量增加,刺激转氨酶发育,次要代谢途径增强,生成苯丙酮酸、苯乙酸和苯乳酸,并从尿中大量排出,故称苯丙酮尿症。苯乳酸使患儿尿液具有特殊的鼠尿臭味。高浓度的Phe及其异常代谢产物抑制酪氨酸酶,使黑色素合成障碍。Phe增高影响脑发育,导致智能发育落后及出现小头畸形、抽搐痉挛等神经系统症状。 PKU的遗传特点是:①患儿父母都是致病基因携带者(杂合子);②患儿从父母各得到一个致病基因,是纯合子;③患儿母亲每次生育有1/4可能为PKU患儿;④近亲结婚的子女发病率较一般人群为高。 人类PAH基因位于第12号染色体上(12q22~12q24.1),PAH基因全长约90kb,有13个外显子和12个内含子,外显子长度在57~892bp之间,成熟mRNA约2.4kb,编码451个氨基酸。内含子长度为1~23kb不等。随着分子生物学技术的发展,北京、上海等地已经开展用单链构型多态性分析(SSCP)、变性梯度凝胶电泳(DGGE)、温度梯度凝胶电泳(TGGE)、点杂交及DNA序列分析等技术对PKU病人进行基因分析,在中国人群中发现了30种以上基因突变(表1),发现外显子7和12的突变占的比例相对较高。其中有一些是中国人特有的突变体,这些基因突变分别导致氨基酸置换、翻译提早终止、mPNA剪切异常、阅读框架移位等。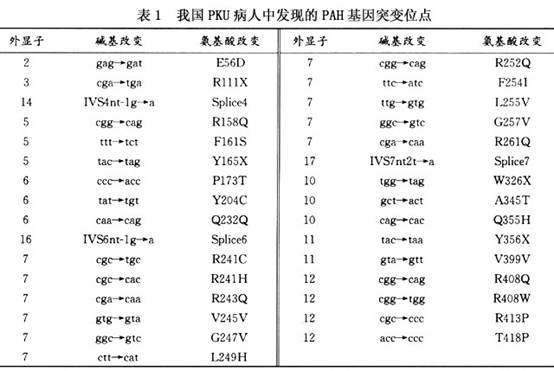 [收起]
苯丙氨酸(phenylalanine,Phe)是人体必需氨基酸,食入体内的Phe一部分用于蛋白质的合成,一部分通过苯丙氨酸羟化酶作用转变为酪氨酸,仅有少量的Phe经过次要的代谢途径在转氨酶的作用下转变成苯丙酮酸。 PKU是因苯丙氨酸羟化酶(phenylalanine hydroxylase,PAH)基因突变导致PAH活性降低或丧失,Phe在肝脏中代谢紊乱所致。PKU患者苯丙氨酸羟化酶缺乏,酪氨酸及正常代谢产物减少,血Phe含量增加,刺激转氨酶发育,次要代谢途径增强,生成苯丙酮酸、苯乙酸和苯乳酸,并从尿中大量排出,故称苯丙酮尿症。苯乳酸使患儿尿液具有特殊的鼠尿臭味。高浓度的Phe及其异...[详细]
[收起]
苯丙氨酸(phenylalanine,Phe)是人体必需氨基酸,食入体内的Phe一部分用于蛋白质的合成,一部分通过苯丙氨酸羟化酶作用转变为酪氨酸,仅有少量的Phe经过次要的代谢途径在转氨酶的作用下转变成苯丙酮酸。 PKU是因苯丙氨酸羟化酶(phenylalanine hydroxylase,PAH)基因突变导致PAH活性降低或丧失,Phe在肝脏中代谢紊乱所致。PKU患者苯丙氨酸羟化酶缺乏,酪氨酸及正常代谢产物减少,血Phe含量增加,刺激转氨酶发育,次要代谢途径增强,生成苯丙酮酸、苯乙酸和苯乳酸,并从尿中大量排出,故称苯丙酮尿症。苯乳酸使患儿尿液具有特殊的鼠尿臭味。高浓度的Phe及其异...[详细]
患儿出生时大多表现正常,新生儿期无明显特殊的临床症状,部分患儿可能出现喂养困难、呕吐、易激惹等非特异性症状。未经治疗的患儿3~4个月后逐渐表现出智力、运动发育落后,头发由黑变黄,皮肤白,全身和尿液有特殊鼠臭味,常有湿疹。 随着年龄增长,患儿智力落后越来越明显,年长儿约60%有严重的智能障碍。2/3患儿有轻微的神经系统体征,如肌张力增高、腱反射亢进、小头畸形等,严重者可有脑性瘫痪。约1/4患儿有癫痫发作,常在18个月以前出现,可表现为婴儿痉挛性发作、点头样发作或其他形式。约80%患儿有脑电图异常,异常表现以癫痫样放电为主,经治疗后血Phe浓度下降,脑电图亦明显改善。PKU患者除了影响智能发育外,可出现一些行为、性格的异常,如忧郁、多动、自卑、孤僻等。 根据不同的临床类型,PKU可分为: 1.经典型PKU(classical PKU) 病儿有典型的临床表现,有程度不等的智能低下,60%属重度低下(IQ低于50)。约1/4病儿有癫痫发作。患者头发、皮肤颜色浅淡,尿液、汗液中散发出鼠臭味,伴有精神行为异常。血Phe浓度>1200μmol/L(20mg/dl),尿FeCl3—和DNPH试验强阳性。 2.中度型PKU(moderate PKU) 临床表现相对较轻,实验室检查结果同经典型PKU,但是血苯丙氨酸在360~1200μmol/L,患儿对治疗反应较好,血苯丙氨酸浓度较经典型患者易控制。 3.轻型PKU(mild PKU) 临床表现较轻或者无症状,血苯丙氨酸小于120~360μmol/L,见于极少数新生儿或早产儿,或者苯丙氨酸羟化酶残余酶活性较高者。 4.四氢生物蝶呤(BH4)缺乏症 临床上将所有血苯丙氨酸>120μmol/L称为高苯丙氨酸血症,从病因上将高苯丙氨酸血症分二大类:苯丙氨酸羟化酶缺乏和PAH的辅酶——四氢生物蝶呤(tetrahydrobiopterin,BH4)缺乏。两类高苯丙氨酸血症治疗方法不同,早期鉴别诊断十分重要。 BH4缺乏症又称非经典型PKU或恶性PKU,由于PAH辅助因子BH4缺乏所致。患儿除了有典型PKU表现外,神经系统表现较为突出,如躯干肌张力下降,四肢肌张力增高,不自主运动,震颤,阵发性角弓反张,顽固性惊厥发作等。BH4缺乏症者单独用低苯丙氨酸饮食治疗可使血苯丙氨酸浓度下降,但神经系统的症状仍呈持续性进展。该病的发生率占PKU的10%左右,因此对所有高苯丙氨酸血症都应进行常规鉴别诊断。CT和MRI检查可见进行性脑萎缩。诊断主要依靠HPLC测定尿中新蝶呤(N)和生物蝶呤(B)。如因6-丙酮酰四氢蝶呤合成酶(PTPS)缺乏时所致的BH4缺乏症,尿中新蝶呤明显增加,N/B增高,B%<10%。如为二氢蝶呤还原酶(DHPR)缺乏时,N正常,B明显增加,N/B降低,B%可增高。三磷酸鸟苷环化水解酶(GTPCH)缺乏者,尿中N和B均非常低,N/B正常。因特异性酶的测定较为复杂困难,可进一步作BH4负荷试验以助诊断。[收起]
患儿出生时大多表现正常,新生儿期无明显特殊的临床症状,部分患儿可能出现喂养困难、呕吐、易激惹等非特异性症状。未经治疗的患儿3~4个月后逐渐表现出智力、运动发育落后,头发由黑变黄,皮肤白,全身和尿液有特殊鼠臭味,常有湿疹。 随着年龄增长,患儿智力落后越来越明显,年长儿约60%有严重的智能障碍。2/3患儿有轻微的神经系统体征,如肌张力增高、腱反射亢进、小头畸形等,严重者可有脑性瘫痪。约1/4患儿有癫痫发作,常在18个月以前出现,可表现为婴儿痉挛性发作、点头样发作或其他形式。约80%患儿有脑电图异常,异常表现以癫痫样放电为主,经治疗后血Phe浓度下降,脑电图亦明显改善。PKU患者除了影响智...[详细]
喂养困难,智力、运动发育落后,常有湿疹,肌张力增高、腱反射亢进,严重者可有脑性瘫痪,癫痫发作,出现忧郁、多动、自卑、孤僻等。
1.新生儿筛查 新生儿期的PKU患儿无任何临床表现,生后3个月后才渐渐出现PKU的表现。随着预防医学科学的发展,苯丙酮尿症的新生儿筛查已逐步成为常规。新生儿筛查即是通过测定血苯丙氨酸,在群体中对每个新生儿进行筛检,使PKU患儿在临床症状尚未出现之前,而其生化等方面的改变已比较明显时得以早期诊断、早期治疗,避免智能落后的发生。 2.尿三氯化铁(FeCl3)及2,4-二硝基苯肼试验(DNPH) (1)三氯化铁(FeCl3)试验:在新鲜尿液5ml加入0.5ml的FeCl3,尿呈绿色为阳性。 (2)2,4-二硝基苯肼试验:在1ml尿液中加入1ml的DNPH试剂,尿液呈黄色荧光反应为阳性。 这两种试验阳性反应也可见于枫糖尿症,胱氨酸血症,故并非为PKU特异性试验,需进一步做血苯丙氨酸测定才能确诊。新生儿PKU因苯丙氨酸代谢旁路尚未健全,患者尿液测定为阴性,该方法不能用于新生儿筛查。 3.血苯丙氨酸测定 有两种方法: (1)Guthrie细菌抑制法:正常浓度<120μmol/L(2mg/dl),PKU>1200μmol/L。 (2)苯丙氨酸荧光定量法:正常值同细菌抑制法。 4.苯丙氨酸负荷试验 对血苯丙氨酸浓度大于正常浓度,<1200μmol/L者口服苯丙氨酸100mg/kg,服前、服后1,2,3,4h分别测定血苯丙氨酸浓度。血苯丙氨酸>1200μmol/L诊断为PKU,<1200μmol/L,为高苯丙氨酸血症。 5.HPLC尿蝶呤图谱分析 10ml晨尿加入0.2g维生素C,酸化尿液后使8cm×10cm新生儿筛查滤纸浸湿,晾干,寄送有条件的实验室分析尿蝶呤图谱,进行四氢生物蝶呤缺乏症的诊断和鉴别诊断。 6.口服四氢生物蝶呤负荷试验 在血Phe浓度>600μmol/L情况下,直接给予口服BH4片20mg/kg,BH4服前,服后2,4,6,8,24h分别取血作Phe测定。对于血Phe浓度<600μmol/L者,可作Phe+BH4联合负荷试验,即给患儿先口服Phe(100mg/kg),服后3h再口服BH4,服Phe前、后1,2,3h,服BH4后2,4,6,8,24h分别采血测Phe浓度。BH4缺乏者,当给予BH4后,因其苯丙氨酸羟化酶活性恢复,血Phe明显下降,PTPS缺乏者,血Phe浓度在服用BH4后4~6h下降至正常;DHPR缺乏者,血Phe浓度一般在服BH4后8h或以后下降至正常;经典型PKU患者因苯丙氨酸羟化酶缺乏,血Phe浓度无明显变化。[收起]
1.新生儿筛查 新生儿期的PKU患儿无任何临床表现,生后3个月后才渐渐出现PKU的表现。随着预防医学科学的发展,苯丙酮尿症的新生儿筛查已逐步成为常规。新生儿筛查即是通过测定血苯丙氨酸,在群体中对每个新生儿进行筛检,使PKU患儿在临床症状尚未出现之前,而其生化等方面的改变已比较明显时得以早期诊断、早期治疗,避免智能落后的发生。 2.尿三氯化铁(FeCl3)及2,4-二硝基苯肼试验(DNPH) (1)三氯化铁(FeCl3)试验:在新鲜尿液5ml加入0.5ml的FeCl3,尿呈绿色为阳性。 (2)2,4-二硝基苯肼试验:在1ml尿液中加入1ml的DNPH试剂,尿液呈黄...[详细]
1.脑电图 约80%病儿有脑电图异常,可表现为高峰节律紊乱、灶性棘波等。 2.CT和MRI检查 患者头颅CT或磁共振影像(MRI)可无异常发现,也可发现有不同程度脑发育不良,表现为脑皮质萎缩和脑白质脱髓鞘病变,后者在MRI的T1加权图像上可显示脑室三角区周围脑组织条形或斑片状高信号区。 3.智力测定 评估智能发育程度。
本病为少数可治性遗传性代谢病之一,应早期确诊和治疗,避免产生神经系统的不可逆性损伤。由于患儿在早期不出现症状,故诊断必须借助实验室检测。在PKU的典型症状出现以后,诊断并不困难,但已为时过晚,因已失去预防脑损伤的时机。必须强调症状前诊断,即在宫内或新生儿早期确诊。我国有些城市正在开展对全部新生儿作PKU普遍筛查,以及时发现所有PKU婴儿。
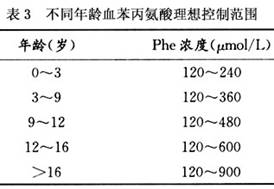
1.治疗原则 PKU是第一种可通过饮食控制治疗的遗传性代谢病。天然食物中均含一定量苯丙氨酸,低蛋白饮食将导致营养不良,因此要用低苯丙氨酸饮食治疗,例如上海生产的华夏2号或其他同类产品,其治疗原则如下: (1)早期治疗:一旦确诊,应立即治疗。开始治疗的年龄越小,预后越好,智能发育可接近正常人。晚治疗者都有程度不等的智能低下。3~5岁后治疗者,可能减轻癫痫和行为异常,但对已存在的严重智能障碍改善不明显。由于新生儿筛查在我国已逐步推广和普及,筛查出的病人往往能在出生1个月内,甚至2周之内得到确诊和治疗,为病儿的健康成长提供了保证。 (2)控制苯丙氨酸摄入:苯丙氨酸是一种必需氨基酸,为生长和体内代谢所必需。PKU患者的智能障碍是由于体内过量的Phe及旁路代谢产物的神经毒性作用而引起,要防止脑损伤,只有减少从食物中摄取苯丙氨酸。血苯丙氨酸应控制在一定范围,以满足其生长发育的需要。一般应保持血苯丙氨酸浓度在120~360μmol/L较为理想。过度治疗将导致苯丙氨酸缺乏,出现嗜睡、厌食、贫血、腹泻,甚至死亡。 (3)调查个体食谱:由于每个患儿对苯丙氨酸的耐受量不同,故在饮食治疗中,仍应根据患儿具体情况调整食谱。低苯丙氨酸奶方治疗至少持续到12岁以上。 (4)家长的合作:是成功的关键因素之一。如果家长充分了解治疗原则,饮食控制得比较合理,病儿的智力发育往往正常。 (5)怀孕前女性患者:成年女性患者在怀孕前应重新开始饮食控制,直到分娩,以免高苯丙氨酸血症影响胎儿。 (6)高危家庭产前诊断:近年来,北京、上海等地都开展了PKU高危家庭产前诊断,通过直接查找基因突变点结合微卫星遗传多态性分析方法(STR,VNTR),成功地对高危家系实施了产前诊断,取得了良好的社会效益。产前诊断之前必须采集PKU患儿及其父母静脉血作家系连锁分析,产前诊断于孕9~12周取绒毛或16~18周取羊水细胞。由于STR多态连锁分析不是直接检测基因突变,因此在应用中必须注意临床诊断的准确性,千万不能将非PAH基因突变的PKU当成PAH突变的病例来进行连锁分析。在产前诊断中还必须严防样品污染,尤其是母体细胞污染。 2.治疗方法 病人一经诊断,应停止给予天然饮食。母乳是婴儿最理想的天然食品,对哺乳期病儿在确诊后虽应暂停母乳喂养,但切勿断奶,以便在控制血苯丙氨酸浓度后能及时添加。 (1)低苯丙氨酸奶方治疗:病人需给予低苯丙氨酸奶方治疗,剂量按每公斤体重需要的蛋白质计算,国产低苯丙氨酸奶方华夏2号的每100g干粉成分见表2。治疗开始1周内,每天服此特殊奶方后2h采血1次,测定血苯丙氨酸浓度。血苯丙氨酸一般在治疗后4天左右降至600μmol/L以下。待血浓度降至理想浓度时(表3),可逐渐少量添加天然饮食,其中首选母乳,因乳中血苯丙氨酸含量仅为牛奶的1/3。较大婴儿及儿童可添加入牛奶、粥、面、蛋等,添加食品应以低蛋白,低苯丙氨酸食物为原则,其量和次数随血苯丙氨酸浓度而定。每位病人能添加的食物种类与量因人而异,与酶的缺陷严重程度有关。较轻病人的血苯丙氨酸浓度较易控制,而严重缺乏者则不容易增添天然食品。每次添加天然饮食或更换食谱后3天,需再复查血苯丙氨酸浓度,以维持血浓度在120~240μmol/L,较为理想。
 (2)生物蝶呤(biopterin,BH4):用于治疗对低苯丙氨酸饮食无反应的变异型,起初所用生物蝶呤(BH4)剂量为每天2.5mg/kg,由于生物蝶呤(BH4)透过血脑屏障甚少,故多数病儿仍不得不合并应用低苯丙氨酸饮食并辅以左旋多巴每天10~15mg/kg,5-羟色胺每天4mg/kg及卡比多巴(Carbidopa)(一种脱羧抑制剂)每天1~2mg/kg。Kapatos等证实用大剂量生物蝶呤(BH4)时,脑组织中可达到有效的治疗浓度,而且鉴于左旋多巴长期使用可导致幻觉、运动障碍和精神症状等不良作用,5-羟色胺也可引起硬皮病样病变。因此尝试单用大剂量BH4治疗此型PKU,最大剂量曾高达每天40mg/kg。由于每一病儿对BH4的敏感程度不同,故对每个病例的治疗剂量应根据以下临床指标予以调整: ①神经系统症状消失与否。 ②血清苯丙氨酸浓度应维持在0.61mmol/L以下。 ③尿中5-羟色胺排出量正常。 单独使用BH4的优点是不需要价格昂贵的特殊饮食治疗,每天仅需服药1次,且不需使用上述可能引起副作用的各种药物。不足之处是并非对每一例本型PKU病儿都具有特殊的治疗效果。如Endres曾报告一例自生后54天开始用生物蝶呤(BH4)治疗的病儿,经治疗随访2年余,效果不理想。为何部分BH4生成不足的病例对此治疗不敏感,机制尚不明确。[收起]
1.治疗原则 PKU是第一种可通过饮食控制治疗的遗传性代谢病。天然食物中均含一定量苯丙氨酸,低蛋白饮食将导致营养不良,因此要用低苯丙氨酸饮食治疗,例如上海生产的华夏2号或其他同类产品,其治疗原则如下: (1)早期治疗:一旦确诊,应立即治疗。开始治疗的年龄越小,预后越好,智能发育可接近正常人。晚治疗者都有程度不等的智能低下。3~5岁后治疗者,可能减轻癫痫和行为异常,但对已存在的严重智能障碍改善不明显。由于新生儿筛查在我国已逐步推广和普及,筛查出的病人往往能在出生1个月内,甚至2周之内得到确诊和治疗,为病儿的健康成长提供了保证。 (2)控制苯丙氨酸摄入:苯丙氨酸是一种必需...[详细]
(2)生物蝶呤(biopterin,BH4):用于治疗对低苯丙氨酸饮食无反应的变异型,起初所用生物蝶呤(BH4)剂量为每天2.5mg/kg,由于生物蝶呤(BH4)透过血脑屏障甚少,故多数病儿仍不得不合并应用低苯丙氨酸饮食并辅以左旋多巴每天10~15mg/kg,5-羟色胺每天4mg/kg及卡比多巴(Carbidopa)(一种脱羧抑制剂)每天1~2mg/kg。Kapatos等证实用大剂量生物蝶呤(BH4)时,脑组织中可达到有效的治疗浓度,而且鉴于左旋多巴长期使用可导致幻觉、运动障碍和精神症状等不良作用,5-羟色胺也可引起硬皮病样病变。因此尝试单用大剂量BH4治疗此型PKU,最大剂量曾高达每天40mg/kg。由于每一病儿对BH4的敏感程度不同,故对每个病例的治疗剂量应根据以下临床指标予以调整: ①神经系统症状消失与否。 ②血清苯丙氨酸浓度应维持在0.61mmol/L以下。 ③尿中5-羟色胺排出量正常。 单独使用BH4的优点是不需要价格昂贵的特殊饮食治疗,每天仅需服药1次,且不需使用上述可能引起副作用的各种药物。不足之处是并非对每一例本型PKU病儿都具有特殊的治疗效果。如Endres曾报告一例自生后54天开始用生物蝶呤(BH4)治疗的病儿,经治疗随访2年余,效果不理想。为何部分BH4生成不足的病例对此治疗不敏感,机制尚不明确。[收起]
1.治疗原则 PKU是第一种可通过饮食控制治疗的遗传性代谢病。天然食物中均含一定量苯丙氨酸,低蛋白饮食将导致营养不良,因此要用低苯丙氨酸饮食治疗,例如上海生产的华夏2号或其他同类产品,其治疗原则如下: (1)早期治疗:一旦确诊,应立即治疗。开始治疗的年龄越小,预后越好,智能发育可接近正常人。晚治疗者都有程度不等的智能低下。3~5岁后治疗者,可能减轻癫痫和行为异常,但对已存在的严重智能障碍改善不明显。由于新生儿筛查在我国已逐步推广和普及,筛查出的病人往往能在出生1个月内,甚至2周之内得到确诊和治疗,为病儿的健康成长提供了保证。 (2)控制苯丙氨酸摄入:苯丙氨酸是一种必需...[详细]
本病为可治性遗传代谢病,早期诊断与治疗,可避免神经系统的不可逆损伤。诊断一旦肯定,应立即给予积极治疗,治疗开始时年龄愈小,效果愈好。
避免亲缘结婚。杂合子之间不应婚配。开展新生儿筛查以早期发现PKU病儿,早期开始治疗,以防止发生智力低下。对于高危家族,可做产前诊断以决定是否作选择性人工流产。对有本病家族史的夫妇必须采用DNA分析或检测羊水中蝶呤等方法对其胎儿进行产前诊断。


