-
科室:
新生儿科
-
别名:
支链酮酸尿症
枫糖尿病
branched-chain ketoaciduria
-
症状:
嗜睡
惊厥
昏迷
-
发病部位:
暂无
-
多发人群:
暂无
-
相关疾病:
氨基酸病
槭糖尿病(maple syrup urine disease)是一种遗传性支链氨基酸代谢障碍的疾病,是由于在细胞线粒体基质内支链α酮酸脱氢酶(BCKD)多酶复合体功能有缺陷。导致这种酶复合体缺陷的病因是编码这一酶复合体中某一成分的基因发生突变。在人体中主要支链氨基酸有亮氨酸、异亮氨酸和缬氨酸。这些支链氨基酸在人体中不能合成,主要从饮食中摄入。像其他氨基酸一样,支链氨基酸可作为蛋白质合成的成分,也可被代谢而产生能量。亮氨酸、异亮氨酸和缬氨酸在降解代谢过程中分别产生α-酮异乙酸,α-酮-1-甲基戊酸和α-酮异戊酸。这3种酮酸进一步代谢则需要BCKD复合物的参与,如果BCKD活性缺乏,则不仅有这3种酮酸在体内堆积;而且使血中3种支链氨基酸浓度升高。3种酮酸对人体神经系统是有毒物质,正是由于其毒性作用而引起枫糖尿病。这3种酮酸从小便排出,使尿呈甜的枫糖浆气味,故而得名。其临床特点是婴儿期喂食困难,中枢神经受损临床表现和代谢性酸中毒,如果未及时得到正确的治疗,常在婴儿期即死亡。[收起]
槭糖尿病(maple syrup urine disease)是一种遗传性支链氨基酸代谢障碍的疾病,是由于在细胞线粒体基质内支链α酮酸脱氢酶(BCKD)多酶复合体功能有缺陷。导致这种酶复合体缺陷的病因是编码这一酶复合体中某一成分的基因发生突变。在人体中主要支链氨基酸有亮氨酸、异亮氨酸和缬氨酸。这些支链氨基酸在人体中不能合成,主要从饮食中摄入。像其他氨基酸一样,支链氨基酸可作为蛋白质合成的成分,也可被代谢而产生能量。亮氨酸、异亮氨酸和缬氨酸在降解代谢过程中分别产生α-酮异乙酸,α-酮-1-甲基戊酸和α-酮异戊酸。这3种酮酸进一步代谢则需要BCKD复合物的参与,如果BCKD活性缺乏,则不仅有这3种...[详细]
本病呈常染色体隐性遗传。由一组支链α酮酸脱氢酶活性缺乏(或酮酸脱羧酶缺乏)或不足所致,分为5型:经典型、间歇型、中间型、维生素B反应型和E3缺乏型。酶活性依次为正常人的0%~2%、2%~40%、5%~25%、20%~40%和5%~10%。患者因呕吐,可造成严重脱水,导致酸中毒。
在哺乳类动物中,BCKD多酶复合体是一种存在于线粒体中的多酶复合体,其功能是催化从支链氨基酸降解而产生的3种支链α酮酸进行氧化性脱羧。酶复合体是围绕一立方形核心。含有24个相同的以双氢脂酰转环酶(dihydrolipoyltranscylase,E2)亚基,通过离子相互作用,而连接在一起,此外还有支链α酮酸脱羧酶(E1)、特异性激酶(E3)和特异性磷酸酶。特异性激酶和磷酸酶通过可逆性磷酸化以调节BCKD复合物活性。E1是一个杂四聚体,由2个E1α和E1β亚基组成。E3为同种二聚体。E1α、E1β、E2和E3的编码基因都可发生突变而导致BCKD多酶复合体活性减低。根据基因突变有人把枫糖尿病分为ⅠA型(E1α亚基突变)、ⅠB型(E1β亚基突变)、Ⅱ型(E2亚基突变),Ⅲ型(E3亚基突变),Ⅳ和Ⅴ则被保留作为特异性激酶和磷酸酶基因突变型,但迄今尚无报道。 本病是一遗传性疾病,其遗传方式为常染色体隐性遗传。遗传缺陷在线粒体中存在的BCKD多酶复合体中的E1、E2和E33个亚基的基因有突变。 BCKD多酶复合体的组成已如前述。E1是焦磷酸硫胺素(TPP)依赖性酶。由2E1α和2E1β形成α2β2四聚体,分子量为170kD。E1α和E1β基因座分别定位在19q和6p,均可发生突变,E1α基因突变则可阻碍其与正常的E1β聚合成四聚体,使E1α迅速被降解,从而使支链α酮酸脱羧酶活性降低或完全丧失,或只形成αβ二聚体,也可形成低分子量的四聚体。Wynn等研究了ⅠA型枫糖尿病病人中E1α与E1β的聚合障碍后指出:在被研究的E1α突变如果是在假定的焦磷酸硫胺素结合袋则对E1亚基的聚合无影响;牵涉到C末端芳香性残基的突变则阻碍亚基聚合动力学和天然的α2β2结构的形成。E1α亚基的突变使支链α-酮酸脱羧酶活性降低或完全丧失。该作者报道E1α突变ⅠA型枫糖尿病表型、聚合状态和特异性活性总结(表1)。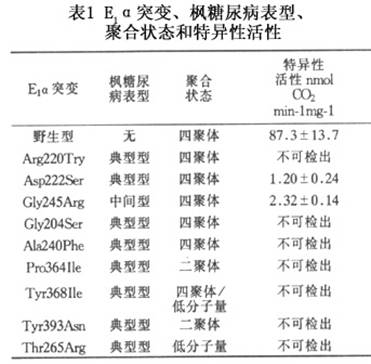 Chinsky等对有BACK活性缺乏的羊水细胞的DNA进行突变分析,显示E1α亚基基因的外显子7有C→T转变,结果E1α亚基有精氨酸R242X的无义突变,即E1α在242位精氨酸位点即停止编码。这种妊娠发生于有多人患枫糖尿病的近亲结婚的后裔中。 E1β亚基也可发生突变。McConnell等报道1例E1β亚基的2个等位基因有不同的突变,其中一个来自母亲的突变基因,在核苷酸编码序列的第526位有A→T突变,导致E1β蛋白有Asp126Tyr取代,这一突变可干扰E1β与辅因子硫胺素之间的相互作用而引起E1β灭活;另一突变的等位基因来自父亲的突变基因。在第970位有C→T突变而形成终止密码子,使E2β蛋白在274位精氨酸位置即被截短。这种突变使BOCK活性小于野生型E1β基因的1%。这种被截短了的E1β变得不稳定。在病人的细胞线粒体中未发现有这种被截短了的E1β蛋白。 E2是24聚体,由载有相同的脂酸(lipoic acid)组成,排列成八面体(octahedral)并在4,3,2点有基团对称。每个E2多肽含有3个独立的折叠区,即载脂酰区(lipoyl-bearing),E1/E3结合区和内核心区。这3个区通过可变通的赘合区连接在一起。这个区在α-酮酸脱氢酶复合体的E2蛋白中是一高度保留区。E2基因座定位于1p31,有11个外显子,长度88kb。文献中已报道的E2基因突变有:插入突变,即在外显子5′端插入17bp;外显子2缺失2个bp,外显子8的5′端的给(donor)位缺失1个bp,结果使外显子8整个缺失;外显子8最后1个核苷酸有G→A突变。其他点突变还有外显子7有T→G突变,导致E2蛋白有Pro 215 Ile取代,外显子6有G→T突变,使E2蛋白在谷氨酸处即终止编码。Chuang等报道由于内在性内含子缺失而引起E2mRNA有失常的剪接者7人。这7例病人的等位基因、BCKD活性、残余酶活性及病人来源(表2)。
Chinsky等对有BACK活性缺乏的羊水细胞的DNA进行突变分析,显示E1α亚基基因的外显子7有C→T转变,结果E1α亚基有精氨酸R242X的无义突变,即E1α在242位精氨酸位点即停止编码。这种妊娠发生于有多人患枫糖尿病的近亲结婚的后裔中。 E1β亚基也可发生突变。McConnell等报道1例E1β亚基的2个等位基因有不同的突变,其中一个来自母亲的突变基因,在核苷酸编码序列的第526位有A→T突变,导致E1β蛋白有Asp126Tyr取代,这一突变可干扰E1β与辅因子硫胺素之间的相互作用而引起E1β灭活;另一突变的等位基因来自父亲的突变基因。在第970位有C→T突变而形成终止密码子,使E2β蛋白在274位精氨酸位置即被截短。这种突变使BOCK活性小于野生型E1β基因的1%。这种被截短了的E1β变得不稳定。在病人的细胞线粒体中未发现有这种被截短了的E1β蛋白。 E2是24聚体,由载有相同的脂酸(lipoic acid)组成,排列成八面体(octahedral)并在4,3,2点有基团对称。每个E2多肽含有3个独立的折叠区,即载脂酰区(lipoyl-bearing),E1/E3结合区和内核心区。这3个区通过可变通的赘合区连接在一起。这个区在α-酮酸脱氢酶复合体的E2蛋白中是一高度保留区。E2基因座定位于1p31,有11个外显子,长度88kb。文献中已报道的E2基因突变有:插入突变,即在外显子5′端插入17bp;外显子2缺失2个bp,外显子8的5′端的给(donor)位缺失1个bp,结果使外显子8整个缺失;外显子8最后1个核苷酸有G→A突变。其他点突变还有外显子7有T→G突变,导致E2蛋白有Pro 215 Ile取代,外显子6有G→T突变,使E2蛋白在谷氨酸处即终止编码。Chuang等报道由于内在性内含子缺失而引起E2mRNA有失常的剪接者7人。这7例病人的等位基因、BCKD活性、残余酶活性及病人来源(表2)。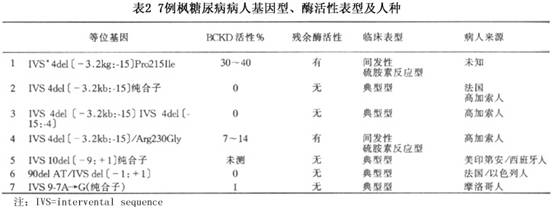 除内含子缺失突变外,还可有其他突变: 1.在内含子8有A→G单个碱基取代而创造了5′端有1个新的剪接位点,使通过激活同一内含子上游的隐性3位点而在外显子8和9之间插入126个核苷酸。预测编码具有包括在氨基末端4个新的氨基酸在内总共只有280个氨基酸的截短的蛋白的mRNA,比具有420个氨基酸的正常E2蛋白少139个氨基酸,即E2蛋白被截短。体外实验表明这种单个碱基取代负责把插入区合并到mRNA中去。 2.在外显子11的第1463位核苷酸有G→T转变而形成终止密码子。这例病人为纯合子,其父母为杂合子。 3.复合性杂合子,在外显子A的第309位有G→A转变,同时在外显子9的1165位有C→G转变,这两种核苷酸突变导致E2蛋白分别有Ile 37 Met和Gly 323 Ser取代。以上3例临床表型为间发型枫糖尿病,其BCKD复合体均有残余功能。 E3是同源二聚体黄色素蛋白(flavoprotein),为所有α-酮酸脱氢酶复合体成员所共有,它是一种特异性激酶,E3基因座定位在7q,也可突变,但比E1和E2少见,E3基因突变除支链α酮酸脱氢酶缺乏外还有丙酮酸脱氢酶和α-酮戊二酸脱氢酶功能受损。 E1、E2和E3中任何一种酶蛋白基因发生突变,都可引起BCKD复合体活性缺乏而导致枫糖尿病的发生。在生化方面主要是支链氨基酸代谢过程中所产生的酮酸在体内堆积而引起频发的严重酮症酸中毒,使神经系统受损。[收起]
在哺乳类动物中,BCKD多酶复合体是一种存在于线粒体中的多酶复合体,其功能是催化从支链氨基酸降解而产生的3种支链α酮酸进行氧化性脱羧。酶复合体是围绕一立方形核心。含有24个相同的以双氢脂酰转环酶(dihydrolipoyltranscylase,E2)亚基,通过离子相互作用,而连接在一起,此外还有支链α酮酸脱羧酶(E1)、特异性激酶(E3)和特异性磷酸酶。特异性激酶和磷酸酶通过可逆性磷酸化以调节BCKD复合物活性。E1是一个杂四聚体,由2个E1α和E1β亚基组成。E3为同种二聚体。E1α、E1β、E2和E3的编码基因都可发生突变而导致BCKD多酶复合体活性减低。根据基因突变有人把枫糖尿病分为Ⅰ...[详细]
除内含子缺失突变外,还可有其他突变: 1.在内含子8有A→G单个碱基取代而创造了5′端有1个新的剪接位点,使通过激活同一内含子上游的隐性3位点而在外显子8和9之间插入126个核苷酸。预测编码具有包括在氨基末端4个新的氨基酸在内总共只有280个氨基酸的截短的蛋白的mRNA,比具有420个氨基酸的正常E2蛋白少139个氨基酸,即E2蛋白被截短。体外实验表明这种单个碱基取代负责把插入区合并到mRNA中去。 2.在外显子11的第1463位核苷酸有G→T转变而形成终止密码子。这例病人为纯合子,其父母为杂合子。 3.复合性杂合子,在外显子A的第309位有G→A转变,同时在外显子9的1165位有C→G转变,这两种核苷酸突变导致E2蛋白分别有Ile 37 Met和Gly 323 Ser取代。以上3例临床表型为间发型枫糖尿病,其BCKD复合体均有残余功能。 E3是同源二聚体黄色素蛋白(flavoprotein),为所有α-酮酸脱氢酶复合体成员所共有,它是一种特异性激酶,E3基因座定位在7q,也可突变,但比E1和E2少见,E3基因突变除支链α酮酸脱氢酶缺乏外还有丙酮酸脱氢酶和α-酮戊二酸脱氢酶功能受损。 E1、E2和E3中任何一种酶蛋白基因发生突变,都可引起BCKD复合体活性缺乏而导致枫糖尿病的发生。在生化方面主要是支链氨基酸代谢过程中所产生的酮酸在体内堆积而引起频发的严重酮症酸中毒,使神经系统受损。[收起]
在哺乳类动物中,BCKD多酶复合体是一种存在于线粒体中的多酶复合体,其功能是催化从支链氨基酸降解而产生的3种支链α酮酸进行氧化性脱羧。酶复合体是围绕一立方形核心。含有24个相同的以双氢脂酰转环酶(dihydrolipoyltranscylase,E2)亚基,通过离子相互作用,而连接在一起,此外还有支链α酮酸脱羧酶(E1)、特异性激酶(E3)和特异性磷酸酶。特异性激酶和磷酸酶通过可逆性磷酸化以调节BCKD复合物活性。E1是一个杂四聚体,由2个E1α和E1β亚基组成。E3为同种二聚体。E1α、E1β、E2和E3的编码基因都可发生突变而导致BCKD多酶复合体活性减低。根据基因突变有人把枫糖尿病分为Ⅰ...[详细]
根据临床表现可将枫糖尿病分为经典型、间歇型、中间型、硫胺素反应型和E3缺乏型,其中以经典型最多见,占75%。中间型和间歇型对硫胺素治疗也有反应。 本病临床表现不均一,主要与BCKD复合体活性降低的程度有关,而BCKD复合体活性是由E1、E2和E3基因的突变所决定。出生后即可发病,患者的尿、汗和耵聍中有特殊的枫糖臭味。临床表现从典型表现到只有轻微症状。 1.经典型(classical type)(新生儿型) 出生后24h内婴儿正常,1周后则出现酮症酸中毒症状,表现为喂食困难、呕吐、代谢性酸中毒及神经系统受损表现。如惊厥、肌张力增高,甚至肌肉强直,呈角弓反张状,也可肌张力增高与松弛交替出现,嗜睡或昏迷。患者可有低血糖,但惊厥和昏迷并非低血糖所致,因为低血糖纠正后,这些症状并无改善。如果未得到正确诊断和治疗,患者常在数周或数月内死亡。本型是枫糖尿病中最严重的,也是最常见的一种类型。即使经治疗而存活,也可有智力低下和神经系统受损的后遗症。此型在Mennonite人群中常见。 2.间歇型(intermittent type) 此型病人常在应激情况下而诱发,如手术、感染和频繁呕吐等。发作时临床表现与经典型相似,并有共济失调,但这型病人BCKD复合体活性残留比典型型者高,8%~10%的病人可接近正常,故症状较轻,严重者也可于发作后死亡。间歇发作时血和尿内支链氨基酸浓度增高,伴低血糖、低钾血症、高氨血症、酮症和酸中毒。MRT检查时T2相双侧苍白球呈高信号改变。 3.中间型(intermediate type) 在新生儿期尿中也有枫糖臭味和轻微症状,以后在患其他疾病时而诱发枫糖尿病。主要是神经系统受累的症状和体征,与经典型相同,但较轻,对用大剂量维生素B1治疗有反应。 4.维生素B(硫胺素)反应型 维生素B1是BCKD复合体的辅酶,当BCKD复合体因E1、E2和E3基因突变而活性降低时,则需要大量以焦磷酸硫胺素为主组成的辅酶。临床表现也较轻,对大剂量(200mg/24h)维生素B1治疗3周才显示出疗效,但也有婴儿患者只要用维生素B1 10mg即有效。 5.二氢脂酰脱氢酶(E3)缺乏型 此型由于BCKD复合体特异性激酶缺乏,这种激酶为所有α-酮酸脱氢酶复合体所共有,故除BCKD复合体活性降低外,还有丙酮酸脱氢酶和α-酮戊二酸脱氢酶功能受损而引起新生儿发生有机酸中毒,患儿出生时正常,后出现全身松弛,肌张力低下,进行性共济失调和严重的神经受损症状和体征,可在儿童期死亡。[收起]
根据临床表现可将枫糖尿病分为经典型、间歇型、中间型、硫胺素反应型和E3缺乏型,其中以经典型最多见,占75%。中间型和间歇型对硫胺素治疗也有反应。 本病临床表现不均一,主要与BCKD复合体活性降低的程度有关,而BCKD复合体活性是由E1、E2和E3基因的突变所决定。出生后即可发病,患者的尿、汗和耵聍中有特殊的枫糖臭味。临床表现从典型表现到只有轻微症状。 1.经典型(classical type)(新生儿型) 出生后24h内婴儿正常,1周后则出现酮症酸中毒症状,表现为喂食困难、呕吐、代谢性酸中毒及神经系统受损表现。如惊厥、肌张力增高,甚至肌肉强直,呈角弓反张状,也可肌张力...[详细]
患者因呕吐,可造成严重脱水,导致酸中毒。应积极治疗原发病及时正确治疗脱水酸中毒。
1.尿液检查 (1)患者尿中由于排出由支链氨基酸代谢所产生的α-酮酸,故有枫糖臭味。Podebrad等对7例病人尿标本用相互选择性多维气相色层析-质谱仪检测法(enautio-MD GC-MS)检查,尿中有臭味的物质为4,5二甲基-3羟-2(5H)-呋喃酮[4,5 dimethyl-3-hydroxy-2(5H)-furanone],又名Sotolone。 (2)测定支链氨基酸(包括亮氨酸、缬氨酸、异亮氨酸和别异亮氨酸):从尿中排出的相应酮酸[即2-酮酸4-甲基-2酮戊酸(2-oxo acid 4-methyl-2-oxopentenoate,KIC)、3-甲基-2-酮丁酸(3-methyl-2-oxobutanoate,KIV)、(S)-(SKMV)、和(R)-3-甲基-2-酮戊酸[(R)-3-methyl-2-oxopantanoate,R-KMV]。Schadewaldt等对10例患典型枫糖尿病的血和尿分别测定其中的各组分浓度,结果表明前述各支链氨基酸从尿中排出的相应代谢产物从低到高依次为KIC(0.1%~25%)、KIV(0.14%~21.3%)、SKMV(0.26%~24.6%)和R-KMV(0.1%~35.9%),尿中排出的游离支链氨基酸则很少。 (3)尿酮酸定性测定:新鲜尿标本加入几滴二硝基苯肼和0.1 NHCl可产生黄色二苯肼沉淀即为阳性。 2.血液检查 (1)血中支链氨基酸测定:用自动氨基酸分析仪或离子交换色层析或串联式质谱荧光分析检测法可直接测定血中支链氨基酸浓度,包括亮氨酸、异亮氨酸、别异亮氨酸(alloisoleucine)和缬氨酸。由于BCKD复合体活性减低或缺如,这些支链氨基酸在血中浓度都升高,其中特别是亮氨酸升高比其他3种支链氨基酸更明显。在正常人血中别异亮氨酸的量很少,在本病中则有升高。因此测定血中别异亮氨酸水平具有诊断意义。 (2)血浆支链氨基酸代谢产物的测定:本病血浆中5-和3-甲基-2酮戊酸对聚体是升高的。 3.血浆氨基酸昼夜变化 本病患者在禁食状态非支链氨基酸由于氨基酸氧化速率比蛋白质裂解速率大,支链氨基酸由于特异性代谢阻断,故支链氨基酸有净得,血液中水平增高。这种在禁食和进食状态下血浆氨基酸的昼夜变化是本病的代谢特征。[收起]
1.尿液检查 (1)患者尿中由于排出由支链氨基酸代谢所产生的α-酮酸,故有枫糖臭味。Podebrad等对7例病人尿标本用相互选择性多维气相色层析-质谱仪检测法(enautio-MD GC-MS)检查,尿中有臭味的物质为4,5二甲基-3羟-2(5H)-呋喃酮[4,5 dimethyl-3-hydroxy-2(5H)-furanone],又名Sotolone。 (2)测定支链氨基酸(包括亮氨酸、缬氨酸、异亮氨酸和别异亮氨酸):从尿中排出的相应酮酸[即2-酮酸4-甲基-2酮戊酸(2-oxo acid 4-methyl-2-oxopentenoate,KIC)、3-甲基-2-酮丁...[详细]
1.CT检查 本病CT检查有脑白质低密度,特别是小脑白质深部、脑干、大脑脚、丘脑和内囊后支。这些神经系统改变在用饮食治疗后有进步,且同时有水肿消失。这一现象可与其他大脑器质性疾病鉴别。 2.做脑MRI检查时可有双侧苍白球在T2相有高信号,但这些CT和MRI的所见并不能作为诊断本病的依据。 3.应用放射性核素技术测定培养的羊水细胞氧化脱羟情况可以进行产前诊断。
本病常发生于婴儿期,且临床表现极不均一,从无症状到有严重临床表现,故诊断不易。如能在新生儿中用串联式分光计进行筛查,则可获得早期诊断。本病虽为遗传性疾病,但遗传方式为常染色体隐性遗传,故家族史对诊断没有帮助。 新生儿尿、汗有枫糖臭味或出现不明原因的代谢性酸中毒应高度怀疑本病。 经典型者多见,主要临床表现为中枢神经受损,如肌张力增加、惊厥、嗜睡和昏迷,同时有代谢性酸中毒。 临床诊断有赖于血浆支链氨基酸及其代谢产物2-酮酸(2-oxo acid)测定都升高,特别是不参与体内蛋白质合成的别异亮氨酸浓度升高更具诊断意义,或测定尿中支链氨基酸的代谢产物也有助于临床诊断。但确诊必须用分子生物技术证实E1、E2或E3有突变,可用周围血中白细胞和皮肤成纤维细胞中所提取出来的这些酶基因的DNA用分子生物学技术进行突变检查。[收起]
本病常发生于婴儿期,且临床表现极不均一,从无症状到有严重临床表现,故诊断不易。如能在新生儿中用串联式分光计进行筛查,则可获得早期诊断。本病虽为遗传性疾病,但遗传方式为常染色体隐性遗传,故家族史对诊断没有帮助。 新生儿尿、汗有枫糖臭味或出现不明原因的代谢性酸中毒应高度怀疑本病。 经典型者多见,主要临床表现为中枢神经受损,如肌张力增加、惊厥、嗜睡和昏迷,同时有代谢性酸中毒。 临床诊断有赖于血浆支链氨基酸及其代谢产物2-酮酸(2-oxo acid)测定都升高,特别是不参与体内蛋白质合成的别异亮氨酸浓度升高更具诊断意义,或测定尿中支链氨基酸的代谢产物也有助于临床诊断...[详细]
本病虽不能根治,但及时正确的治疗可使患儿存活,症状可得到改善。对代谢失代偿的急性危象的治疗要采取紧急措施,否则患儿易于夭折,治疗方法如下。 1.氨基酸与营养治疗 静脉滴注或从鼻胃管滴入特殊配制的无肝用氨基酸输液(支链氨基酸)的混合性氨基酸溶液,同时从静脉输给葡萄糖(或高张葡萄糖)和电解质。在治疗前患儿血浆亮氨酸水平一般大于40mg/dl。用前述治疗,血浆亮氨酸可迅速下降。从鼻胃管滴入与静脉滴注有同样效果。 急性危象治疗的目的是要改善神经系统的不良结局,要达到此目的在于:①缩短神志改变的时间和严重程度;②尽可能地使血浆亮氨酸水平降低。这是治疗急性危象的两条重要原则。危象缓解后,本病患者应长期少吃或禁食含肝用氨基酸输液(支链氨基酸)的食物。本病妇女怀孕时,在怀孕过程中血浆支链氨基酸水平如能保持在100~300mmol/L则可分娩正常婴儿。从妊娠第22周以后,亮氨酸耐受性从350mg/d进行性增高到2100mg/d。分娩后应仔细监测血中支链氨基酸水平,以减少产褥期发生代谢失代偿的危险。 2.透析治疗 此种治疗是通过透析以清除堆积在血中的大量亮氨酸,可用于抢救急危重患者。Jouvet等对3例有生命危险的新生儿采用了3种不同的治疗方法:即静脉-静脉持续血液过滤(haemofiltration)、血液透析过滤(haemodiafiltration)和血液透析。通过治疗3例新生儿血浆中的亮氨酸分别为2186,3818和2536mmol/L急剧地下降到1131,1275和488mmol/L,患儿的神经系统状态迅速得到改善。在月龄分别为22,13和11时,3例患儿均有正常的智商。患儿对这些治疗耐受良好,副作用有低温和血球压积下降。 3.交换输血 此方法是将患者的血输给正常人,将正常人的血输给患者。正常人有处理支链氨基酸的能力,患者的血输给正常人无不良反应和后果。而患者血中亮氨酸水平可降低。 4.大剂量维生素B1(硫胺素)和静脉高能营养 对维生素B1(硫胺素)治疗有反应的病型主要是Ⅱ型枫糖尿病(即E2基因突变),但中间型和轻型病人也有反应。对维生素B1(硫胺素)治疗有反应的前提是E1正常。因为当E2与E1形成复合物时使E2酶对焦磷酸硫胺素的亲和力增高,同时焦磷酸硫胺素可使BCKD复合体保持稳定,由此使病人对支链氨基酸的耐受性增加。维生素B1(硫胺素)剂量一般要大,100mg/d;但也有用1Omg取得疗效者。在用大剂量维生素B1(硫胺素)治疗的同时要静脉补充高能营养,供给充足的热卡、维生素、水和电解质来满足需要。 5.肝脏移植 至今只有3例作了肝脏移植,移植后全身支链2-氧酸脱氢酶活性明显升高,不需限制饮食,在分解代谢增高的情况下,没有代谢失代偿的危险。但考虑到所需费用,手术风险和结果,Wendel等认为肝移植并不比经典的限制饮食治疗更好。[收起]
本病虽不能根治,但及时正确的治疗可使患儿存活,症状可得到改善。对代谢失代偿的急性危象的治疗要采取紧急措施,否则患儿易于夭折,治疗方法如下。 1.氨基酸与营养治疗 静脉滴注或从鼻胃管滴入特殊配制的无肝用氨基酸输液(支链氨基酸)的混合性氨基酸溶液,同时从静脉输给葡萄糖(或高张葡萄糖)和电解质。在治疗前患儿血浆亮氨酸水平一般大于40mg/dl。用前述治疗,血浆亮氨酸可迅速下降。从鼻胃管滴入与静脉滴注有同样效果。 急性危象治疗的目的是要改善神经系统的不良结局,要达到此目的在于:①缩短神志改变的时间和严重程度;②尽可能地使血浆亮氨酸水平降低。这是治疗急性危象的两条重要原则。危象缓解...[详细]
预后取决于及时诊断和及时治疗,特别是急性危象期。如果抢救不及时,患病婴儿极易死亡;如果得到及时治疗,患者神志很快恢复可无神经系统后遗症。 患儿的长期预后有待观望。在应激状况下如感染或手术时可能发生严重的酮酸症、脑水肿甚至死亡。常见的后遗症是智力缺陷和神经系统损害。
本病虽为遗传性疾病,但遗传方式为常染色体隐性遗传,故家族史对诊断没有帮助。


